Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines

 ,
, 

Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. SAM Synthesis
2.3. Preparation of SAM Formulations
2.3.1. Formulation of Liposome, Solid-lipid Nanoparticles and Polymeric Nanoparticles
2.3.2. Formulation of Emulsions
2.3.3. Preparation of SAM-Loaded Formulations
2.4. Quantification of SAM Loading and Adsorption Efficiency
2.5. Physiochemical Characterization of Formulations
2.6. RNase Protection Assay
2.7. Cell Toxicity Assay in Baby Hamster Kidney Cells
2.8. Cellular Association Efficiency in Baby Hamster Kidney Cells
2.9. In Vitro Potency of SAM-RVG Formulations
2.10. Evaluation of Immune Responses in Vivo of Different Selected Adjuvants and Their Associated Antigen
2.10.1. Determination of Antigen-Specific Serum Antibody Titers by Enzyme-Linked Immunosorbent Assay (ELISA)
2.10.2. Intracellular Cytokine Staining (ICS) in Splenocytes
2.11. Statistical Analysis
3. Results
3.1. Evaluation of Physical Attributes of Liposomes, Solid Lipid Particles, Polymeric Particles and Emulsions Produced with SAM-RVG
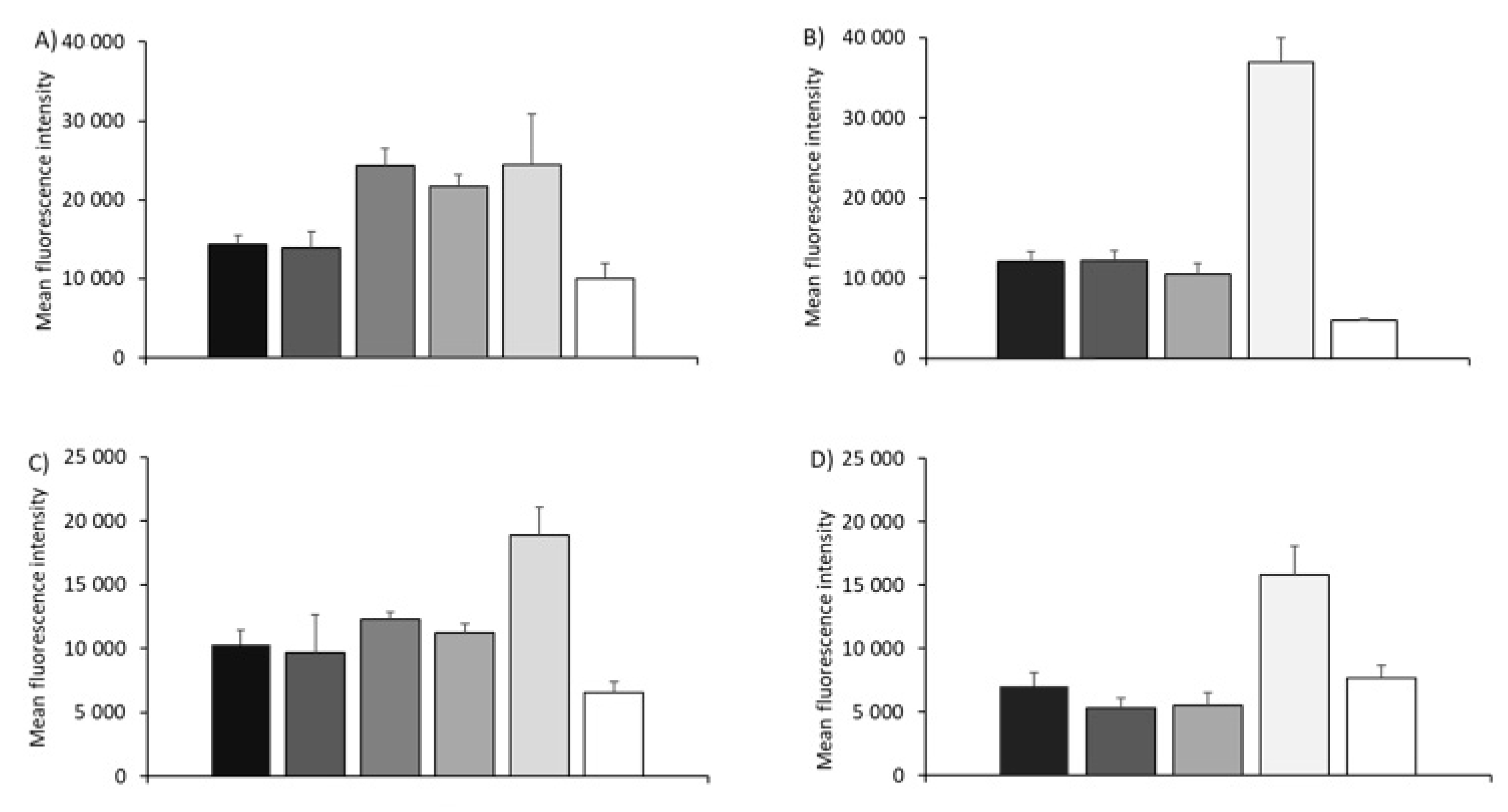
3.2. Cell Association of SAM-RVG-Loaded Liposomes, SLNs, NPs and Emulsions in BHK Cell Line
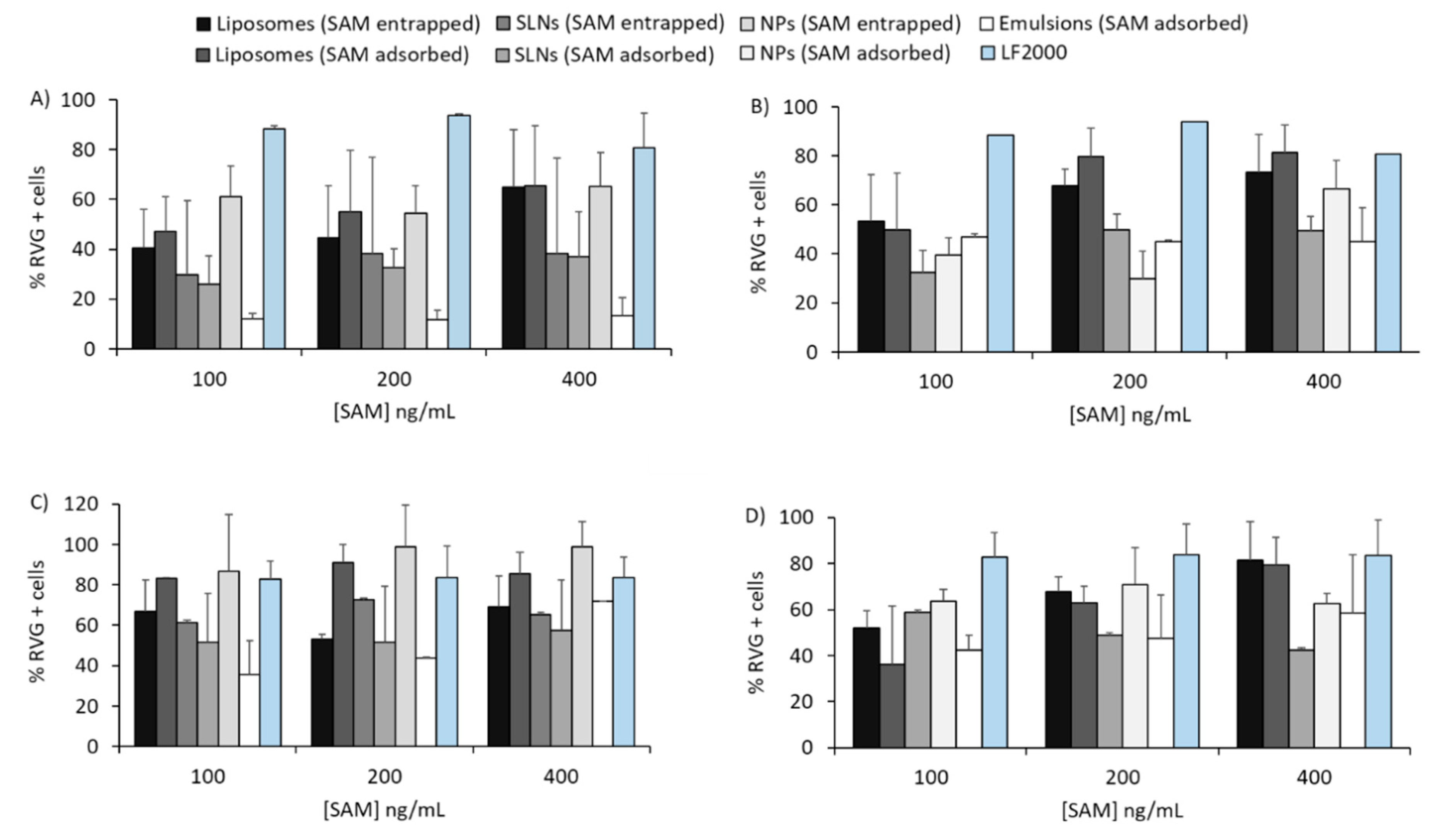
3.3. In Vitro Potency Assay with SAM-RVG Formulations
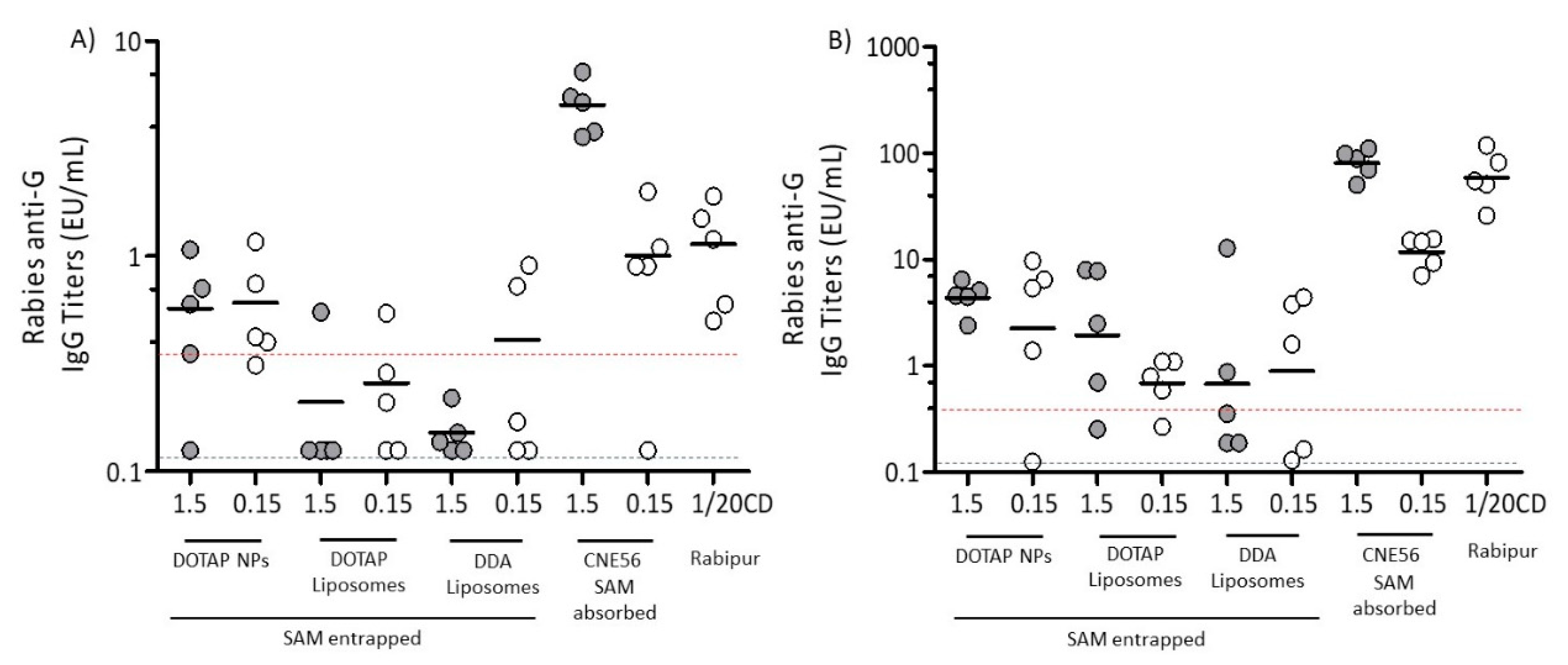
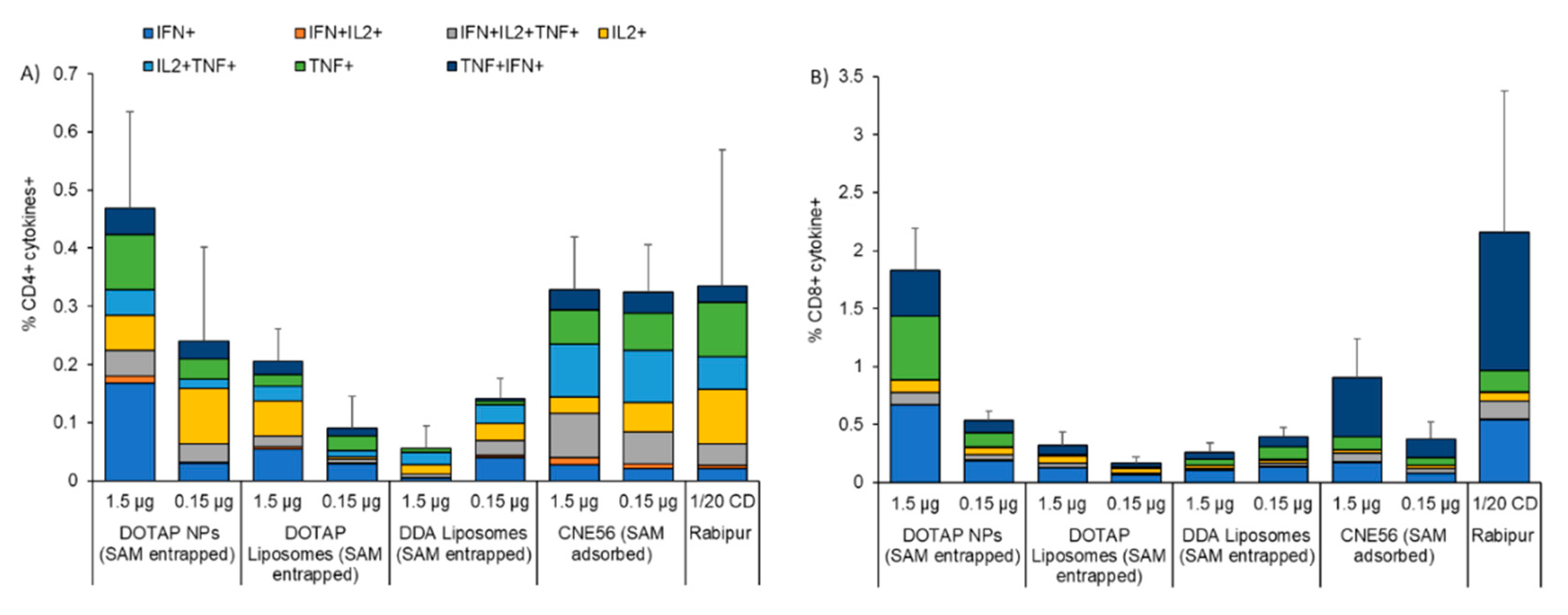
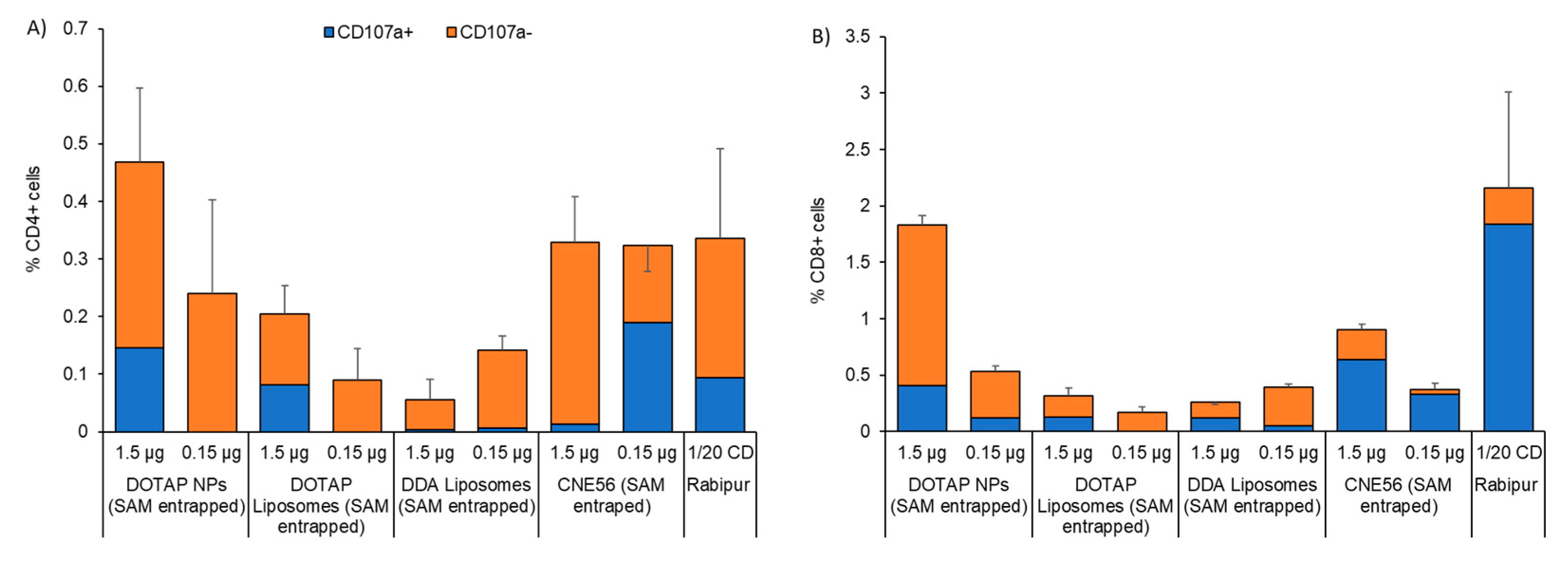
3.4. Immunogenicity of Different Vaccine Candidates Encoding RVG after Intramuscular Injection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.P.; Zabielski, M.A.; Schmaltz, F.L.; Brownlee, G.G.; Bussey, L.A.; Marshall, K.; Borralho, T.; Nagata, L.P. DNA vaccination against respiratory influenza virus infection. Vaccine 2001, 19, 2461–2467. [Google Scholar] [CrossRef]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Luo, D.; Saltzman, W.M. Synthetic DNA delivery systems. Nat. Biotechnol. 2000, 18, 33–37. [Google Scholar] [CrossRef]
- Kallen, K.-J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology: Self-adjuvanted RNActive(®) vaccines. Hum. Vaccines Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef]
- Chatterjee, S.; Pal, J.K. Role of 5’-and 3’-untranslated regions of mRNAs in human diseases. Biol. Cell 2009, 101, 251–262. [Google Scholar] [CrossRef]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Self-amplifying mRNA vaccines. Adv. Genet. 2015, 89, 179–233. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Nayerossadat, N.; Maedeh, T.; Ali, P.A. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Gehl, J. Electroporation: Theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol. Scand. 2003, 177, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.D.; Li, S. Electroporation advances in large animals. Curr. Gene Ther. 2009, 9, 316–326. [Google Scholar] [CrossRef]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Otten, G.R.; Brazzoli, M.; Buccato, S.; Bonci, A.; et al. Rapidly produced SAM(®) vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microb. Infect. 2013, 2, e52. [Google Scholar] [CrossRef]
- Magini, D.; Giovani, C.; Mangiavacchi, S.; Maccari, S.; Cecchi, R.; Ulmer, J.B.; De Gregorio, E.; Geall, A.J.; Brazzoli, M.; Bertholet, S. Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLoS ONE 2016, 11, e0161193. [Google Scholar] [CrossRef]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Kotelevtsev, Y.V.; Koteliansky, V. Lipid nanoparticles for targeted siRNA delivery—Going from bench to bedside. Int. J. Nanomed. 2016, 11, 3077–3086. [Google Scholar] [CrossRef]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8529–8533. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Frederik, P.M.; Gregoriadis, G. Liposome-mediated DNA vaccination: The effect of vesicle composition. Vaccine 2001, 19, 3301–3310. [Google Scholar] [CrossRef]
- Walker, C.; Selby, M.; Erickson, A.; Cataldo, D.; Valensi, J.P.; Van Nest, G.V. Cationic lipids direct a viral glycoprotein into the class I major histocompatibility complex antigen-presentation pathway. Proc. Natl. Acad. Sci. USA 1992, 89, 7915–7918. [Google Scholar] [CrossRef] [PubMed]
- Agger, E.M. Novel adjuvant formulations for delivery of anti-tuberculosis vaccine candidates. Adv. Drug Deliv. Rev. 2016, 102, 73–82. [Google Scholar] [CrossRef]
- Agger, E.M.; Rosenkrands, I.; Hansen, J.; Brahimi, K.; Vandahl, B.S.; Aagaard, C.; Werninghaus, K.; Kirschning, C.; Lang, R.; Christensen, D.; et al. Cationic Liposomes Formulated with Synthetic Mycobacterial Cordfactor (CAF01): A Versatile Adjuvant for Vaccines with Different Immunological Requirements. PLoS ONE 2008, 3, e3116. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal Formulations in Clinical Use: An Updated Review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- Barba, A.A.; Bochicchio, S.; Dalmoro, A.; Lamberti, G. Lipid Delivery Systems for Nucleic-Acid-Based-Drugs: From Production to Clinical Applications. Pharmaceutics 2019, 11, 360. [Google Scholar] [CrossRef]
- Xiao, Y.; Shi, K.; Qu, Y.; Chu, B.; Qian, Z. Engineering Nanoparticles for Targeted Delivery of Nucleic Acid Therapeutics in Tumor. Mol. Ther. Methods Clin. Dev. 2019, 12, 1–18. [Google Scholar] [CrossRef]
- Miyata, K.; Uchida, S.; Naito, M.; Kataoka, K. Polymer nanotechnology for nucleic acid delivery. Drug Deliv. Syst. 2016, 31, 44–53. [Google Scholar] [CrossRef][Green Version]
- Templeton, N. Liposomal Delivery of Nucleic Acids In Vivo. DNA Cell Biol. 2003, 21, 857–867. [Google Scholar] [CrossRef]
- Truong, N.P.; Gu, W.; Prasadam, I.; Jia, Z.; Crawford, R.; Xiao, Y.; Monteiro, M.J. An influenza virus-inspired polymer system for the timed release of siRNA. Nat. Commun. 2013, 4, 1902. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Hussain, M.T.; Roces, C.B.; Anderluzzi, G.; Kastner, E.; Salmaso, S.; Kirby, D.J.; Perrie, Y. Microfluidics based manufacture of liposomes simultaneously entrapping hydrophilic and lipophilic drugs. Int. J. Pharm. 2016, 514, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Kastner, E.; Verma, V.; Lowry, D.; Perrie, Y. Microfluidic-controlled manufacture of liposomes for the solubilisation of a poorly water soluble drug. Int. J. Pharm. 2015, 485, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Anderluzzi, G.; Perrie, Y. Microfluidic Manufacture of Solid Lipid Nanoparticles: A Case Study on Tristearin-Based Systems. Drug Deliv. Lett. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Barnadas-Rodríguez, R.; Sabés, M. Factors involved in the production of liposomes with a high-pressure homogenizer. Int. J. Pharm. 2001, 213, 175–186. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Blakney, A.K.; McKay, P.F.; Yus, B.I.; Aldon, Y.; Shattock, R.J. Inside out: Optimization of lipid nanoparticle formulations for exterior complexation and in vivo delivery of saRNA. Gene Ther. 2019, 26, 363–372. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef]
- Brazzoli, M.; Magini, D.; Bonci, A.; Buccato, S.; Giovani, C.; Kratzer, R.; Zurli, V.; Mangiavacchi, S.; Casini, D.; Brito, L.M.; et al. Induction of Broad-Based Immunity and Protective Efficacy by Self-amplifying mRNA Vaccines Encoding Influenza Virus Hemagglutinin. J. Virol. 2015, 90, 332–344. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Feyssaguet, M.; Dacheux, L.; Audry, L.; Compoint, A.; Morize, J.L.; Blanchard, I.; Bourhy, H. Multicenter comparative study of a new Elisa, Platelia Rabies II, for the detection and titration of anti-rabies glycoprotein antibodies and comparison with the rapid fluorescent focus inhibition test (RFFIT) on human samples from vaccinated and non-vaccinated people. Vaccine 2007, 25, 2244–2251. [Google Scholar] [CrossRef] [PubMed]
- Stantic-Pavlinic, M.; Hostnik, P.; Levicnik-Stezinar, S.; Lijana, Z. Vaccination against rabies and protective antibodies—Comparison of ELISA and fluorescent antibody virus neutralization (FAVN) assays. Vet. Arch. 2006, 76, 281–289. [Google Scholar]
- World Health Organization. Expert Consultation on Rabies; WHO: Geneva, Switzerland, 2005; Volume 931, pp. 1–88. [Google Scholar]
- Zaritskaya, L.; Shurin, M.R.; Sayers, T.J.; Malyguine, A.M. New flow cytometric assays for monitoring cell-mediated cytotoxicity. Exp. Rev. Vaccines 2010, 9, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Geall, A.; Settembre, E.C.; Ulmer, J.B. Using self-amplifying mRNA vaccines to facilitate a rapid response to pandemic influenza. Eur. Pharm. Rev. 2014, 19, 20–23. [Google Scholar]
- Perrie, Y.; Gregoriadis, G. Liposome-entrapped plasmid DNA: Characterisation studies. Biochim. Biophys. Acta 2000, 1475, 125–132. [Google Scholar] [CrossRef]
- Bochicchio, S.; Dalmoro, A.; Barba, A.A.; Grassi, G.; Lamberti, G. Liposomes as siRNA delivery vectors. Curr. Drug Metab. 2014, 15, 882–892. [Google Scholar] [CrossRef]
- Fisher, R.K.; Mattern-Schain, S.I.; Best, M.D.; Kirkpatrick, S.S.; Freeman, M.B.; Grandas, O.H.; Mountain, D.J.H. Improving the efficacy of liposome-mediated vascular gene therapy via lipid surface modifications. J. Surg. Res. 2017, 219, 136–144. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Nicolau, C.; Le Pape, A.; Soriano, P.; Fargette, F.; Juhel, M.F. In vivo expression of rat insulin after intravenous administration of the liposome-entrapped gene for rat insulin I. Proc Natl Acad Sci USA 1983, 80, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef]
- Bruxel, F.; Cojean, S.; Bochot, A.; Teixeira, H.; Bories, C.; Loiseau, P.M.; Fattal, E. Cationic nanoemulsion as a delivery system for oligonucleotides targeting malarial topoisomerase II. Int. J. Pharm. 2011, 416, 402–409. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Tsai, T.F.; Brito, L.A. Emulsion based vaccine adjuvants. Hum. Vaccines Immunother. 2013, 9, 1698–1700. [Google Scholar] [CrossRef]
- Ott, G.; Singh, M.; Kazzaz, J.; Briones, M.; Soenawan, E.; Ugozzoli, M.; O’Hagan, D.T. A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J. Control. Release 2002, 79, 1–5. [Google Scholar] [CrossRef]
- Borchard, G. Chitosans for gene delivery. Adv. Drug Deliv. Rev. 2001, 52, 145–150. [Google Scholar] [CrossRef]
- Florea, B.; Meaney, C.; Junginger, H.E.; Borchard, G. Transfection efficiency and toxicity of polyethylenimine in differentiated Calu-3 and nondifferentiated COS-1 cell cultures. AAPS J. 2002, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Anderluzzi, G.; Lou, G.; Su, Y.; Perrie, Y. Scalable Manufacturing Processes for Solid Lipid Nanoparticles. Pharm. Nanotechnol. 2019, 7, 444–459. [Google Scholar] [CrossRef]
- Bhalekar, M.; Upadhaya, P.; Madgulkar, A. Formulation and characterization of solid lipid nanoparticles for an anti-retroviral drug darunavir. Appl. Nanosci. 2017, 7, 47–57. [Google Scholar] [CrossRef]
- Weiss, J.; Decker, E.; McClements, D.; Kristbergsson, K.; Helgason, T.; Awad, T. Solid Lipid Nanoparticles as Delivery Systems for Bioactive Food Components. Food Biophys. 2008, 3, 146–154. [Google Scholar] [CrossRef]
- Montana, G.; Bondi, M.L.; Carrotta, R.; Picone, P.; Craparo, E.F.; San Biagio, P.L.; Giammona, G.; Di Carlo, M. Employment of cationic solid-lipid nanoparticles as RNA carriers. Bioconjug. Chem. 2007, 18, 302–308. [Google Scholar] [CrossRef]
- De Jesus, M.B.; Zuhorn, I.S. Solid lipid nanoparticles as nucleic acid delivery system: Properties and molecular mechanisms. J. Control. Release 2015, 201, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Korsholm, K.S.; Agger, E.M.; Foged, C.; Christensen, D.; Dietrich, J.; Andersen, C.S.; Geisler, C.; Andersen, P. The adjuvant mechanism of cationic dimethyldioctadecylammonium liposomes. Immunology 2007, 121, 216–226. [Google Scholar] [CrossRef] [PubMed]
- De Serrano, L.O.; Burkhart, D.J. Liposomal vaccine formulations as prophylactic agents: Design considerations for modern vaccines. J. Nanobiotechnol. 2017, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.P.; Demeneix, B.; Loeffler, J.P.; Perez-Mutul, J. Efficient gene transfer into mammalian primary endocrine cells with lipopolyamine-coated DNA. Proc. Natl. Acad. Sci. USA 1989, 86, 6982–6986. [Google Scholar] [CrossRef] [PubMed]
- Kopatz, I.; Remy, J.S.; Behr, J.P. A model for non-viral gene delivery: Through syndecan adhesion molecules and powered by actin. J. Gene Med. 2004, 6, 769–776. [Google Scholar] [CrossRef]
- Mislick, K.A.; Baldeschwieler, J.D. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 12349–12354. [Google Scholar] [CrossRef]
- Hafez, I.M.; Ansell, S.; Cullis, P.R. Tunable pH-sensitive liposomes composed of mixtures of cationic and anionic lipids. Biophys. J. 2000, 79, 1438–1446. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Christensen, D.; Bramwell, V.W.; Lindenstrøm, T.; Agger, E.M.; Andersen, P.; Perrie, Y. Liposomal cationic charge and antigen adsorption are important properties for the efficient deposition of antigen at the injection site and ability of the vaccine to induce a CMI response. J. Control. Release Off. J. Control. Release Soc. 2010, 145, 102–108. [Google Scholar] [CrossRef]
- Iavarone, C.; O’hagan, D.T.; Yu, D.; Delahaye, N.F.; Ulmer, J.B. Mechanism of action of mRNA-based vaccines. Exp. Rev. Vaccines 2017, 16, 871–881. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Maitani, Y.; Igarashi, S.; Sato, M.; Hattori, Y. Cationic liposome (DC-Chol/DOPE = 1:2) and a modified ethanol injection method to prepare liposomes, increased gene expression. Int. J. Pharm. 2007, 342, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.W.; Langner, M.; Zhao, Y.L.; Ross, P.; Hurley, E.; Chan, K. The role of helper lipids in cationic liposome-mediated gene transfer. Biophys. J. 1996, 71, 590–599. [Google Scholar] [CrossRef]
- Luten, J.; van Nostrum, C.F.; De Smedt, S.C.; Hennink, W.E. Biodegradable polymers as non-viral carriers for plasmid DNA delivery. J. Control. Release 2008, 126, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Zhou, W.Z.; Prabha, S.; Sahoo, S.K.; Labhasetwar, V. Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: Implications for drug and gene delivery. FASEB J. 2002, 16, 1217–1226. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ghosh, R.N.; Maxfield, F.R. Endocytosis. Physiol. Rev. 1997, 77, 759–803. [Google Scholar] [CrossRef]
- Roces, C.B.; Hussain, M.T.; Schmidt, S.T.; Christensen, D.; Perrie, Y. Investigating Prime-Pull Vaccination through a Combination of Parenteral Vaccination and Intranasal Boosting. Vaccines 2019, 8, 10. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.; Coco, R.; Breton, A.; Preat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release Off. J. Control. Release Soc. 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P. An Overview of Poly(lactic-co-glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater. Sci. Eng. C. 2018, 92, 1041–1060. [Google Scholar] [CrossRef]
- Xu, H.; Kona, S.; Su, L.-C.; Tsai, Y.-T.; Dong, J.-F.; Brilakis, E.S.; Tang, L.; Banerjee, S.; Nguyen, K.T. Multi-ligand poly(L-lactic-co-glycolic acid) nanoparticles inhibit activation of endothelial cells. J. Cardiovasc. Transl. Res. 2013, 6, 570–578. [Google Scholar] [CrossRef]
- Nguyen, D.N.; Green, J.J.; Chan, J.M.; Longer, R.; Anderson, D.G. Polymeric Materials for Gene Delivery and DNA Vaccination. Adv. Mater. 2009, 21, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Safaiyan, S.; Rafati, S. Improvement of different vaccine delivery systems for cancer therapy. Mol. Cancer 2011, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- McNeil, S.E.; Vangala, A.; Bramwell, V.W.; Hanson, P.J.; Perrie, Y. Lipoplexes formulation and optimisation: In vitro transfection studies reveal no correlation with in vivo vaccination studies. Curr. Drug. Deliv. 2010, 7, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.; Agger, E.M.; Andreasen, L.V.; Kirby, D.; Andersen, P.; Perrie, Y. Liposome-based cationic adjuvant formulations (CAF): Past, present, and future. J. Liposome Res. 2009, 19, 2–11. [Google Scholar] [CrossRef]
- Tandrup Schmidt, S.; Foged, C.; Korsholm, K.S.; Rades, T.; Christensen, D. Liposome-Based Adjuvants for Subunit Vaccines: Formulation Strategies for Subunit Antigens and Immunostimulators. Pharmaceutics 2016, 8, 7. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Bramwell, V.W.; Christensen, D.; Agger, E.M.; Andersen, P.; Perrie, Y. Liposomes based on dimethyldioctadecylammonium promote a depot effect and enhance immunogenicity of soluble antigen. J. Control. Release 2010, 142, 180–186. [Google Scholar] [CrossRef]
- Henriksen-Lacey, M.; Devitt, A.; Perrie, Y. The vesicle size of DDA:TDB liposomal adjuvants plays a role in the cell-mediated immune response but has no significant effect on antibody production. J. Control. Release 2011, 154, 131–137. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef]
- Pepini, T.; Pulichino, A.M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef]
- Durymanov, M.; Reineke, J. Non-viral Delivery of Nucleic Acids: Insight Into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Hemann, E.A.; McGill, J.L.; Legge, K.L. Chronic ethanol exposure selectively inhibits the influenza-specific CD8 T cell response during influenza a virus infection. Alcohol. Clin. Exp. Res. 2014, 38, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.S.; Cao, M.; Wong, W.W.; Fischer, K.P.; Addison, W.R.; Kwon, G.S.; Tyrrell, D.L.; Samuel, J. Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. J. Control. Release 2005, 102, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.E.; Titball, R.; Williamson, D. Vaccine delivery using nanoparticles. Front. Cell Infect. Microbiol. 2013, 3, 13. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation Type | DOTAP | DDA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Size (d.nm) | PDI | ZP (mV) | EE/AE (%) | Size (d.nm) | PDI | ZP (mV) | EE/AE (%) | ||
| Liposomes | No SAM | 42 ± 4 | 0.25 ± 0.01 | 58 ± 3 | - | 39 ± 6 | 0.20 ± 0.03 | 45 ± 7 | - |
| Adsorbed | 118 ± 25 | 0.36 ± 0.20 | 39 ± 5 | 97 ± 1 | 186 ± 15 | 0.16 ± 0.02 | 27 ± 4 | 99 ± 0.1 | |
| Entrapped | 85 ± 5 | 0.17 ± 0.02 | 27 ± 3 | 96 ± 1 | 196 ± 9 | 0.21 ± 0.02 | 43 ± 3 | 99 ± 0.2 | |
| SLNs | No SAM | 64 ± 1 | 0.11 ± 0.01 | 30 ± 4 | - | 70 ± 6 | 0.26 ± 0.02 | 46 ± 4 | - |
| Adsorbed | 120 ± 2 | 0.21 ± 0.02 | 15 ± 2 | 97 ± 1 | 201 ± 62 | 0.29 ± 0.04 | 26 ± 4 | 99 ± 0.1 | |
| Entrapped | 187 ± 17 | 0.14 ± 0.01 | 23 ± 1 | 98 ± 1 | - | - | - | - | |
| NPs | No SAM | 39 ± 9 | 0.13 ± 0.07 | 49 ± 6 | - | 58 ± 3 | 0.07 ± 0.02 | 38 ± 5 | - |
| Adsorbed | - | - | - | - | 267 ± 34 | 0.27 ± 0.05 | 26 ± 4 | 99 ± 0.1 | |
| Entrapped | 198 ± 6 | 0.23 ± 0.02 | 26 ± 6 | 98 ± 1 | - | - | - | - | |
| Emulsions | No SAM | 150 ± 7 | 0.05 ± 0.01 | 38 ± 2 | - | 196 ± 24 | 0.19 ± 0.07 | 38 ± 3 | - |
| Adsorbed | 182 ± 33 | 0.19 ± 0.01 | 35 ± 2 | 95 ± 2 | 209 ± 9 | 0.08 ± 0.03 | 35 ± 3 | 90 ± 0.5 | |
| Entrapped | - | - | - | - | - | - | - | - | |
| Progression Selection Criteria | ||||
|---|---|---|---|---|
| Cationic Lipid | Delivery Platform | Method of Association | Progress to In Vitro Based on Physio-Chemical Attributes | Progress to In Vitro Based on In Vitro Efficacy |
| DOTAP | Liposomes | Adsorbed | ✓ | ✕ |
| Entrapped | ✓ | ✓ | ||
| SLNs | Adsorbed | ✓ | ✕ | |
| Entrapped | ✓ | ✕ | ||
| NPs | Adsorbed | ✕ | ✕ | |
| Entrapped | ✓ | ✓ | ||
| Emulsions | Adsorbed | ✓ | ✕ | |
| DDA | Liposomes | Adsorbed | ✓ | ✕ |
| Entrapped | ✓ | ✓ | ||
| SLNs | Adsorbed | ✓ | ✕ | |
| Entrapped | ✕ | ✕ | ||
| NPs | Adsorbed | ✓ | ✕ | |
| Entrapped | ✕ | ✕ | ||
| Emulsions | Adsorbed | ✓ | ✕ | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderluzzi, G.; Lou, G.; Gallorini, S.; Brazzoli, M.; Johnson, R.; O’Hagan, D.T.; Baudner, B.C.; Perrie, Y. Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines. Vaccines 2020, 8, 212. https://doi.org/10.3390/vaccines8020212
Anderluzzi G, Lou G, Gallorini S, Brazzoli M, Johnson R, O’Hagan DT, Baudner BC, Perrie Y. Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines. Vaccines. 2020; 8(2):212. https://doi.org/10.3390/vaccines8020212
Chicago/Turabian StyleAnderluzzi, Giulia, Gustavo Lou, Simona Gallorini, Michela Brazzoli, Russell Johnson, Derek T. O’Hagan, Barbara C. Baudner, and Yvonne Perrie. 2020. "Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines" Vaccines 8, no. 2: 212. https://doi.org/10.3390/vaccines8020212
APA StyleAnderluzzi, G., Lou, G., Gallorini, S., Brazzoli, M., Johnson, R., O’Hagan, D. T., Baudner, B. C., & Perrie, Y. (2020). Investigating the Impact of Delivery System Design on the Efficacy of Self-Amplifying RNA Vaccines. Vaccines, 8(2), 212. https://doi.org/10.3390/vaccines8020212