Modified Alphavirus-Vesiculovirus Hybrid Vaccine Vectors for Homologous Prime-Boost Immunotherapy of Chronic Hepatitis B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
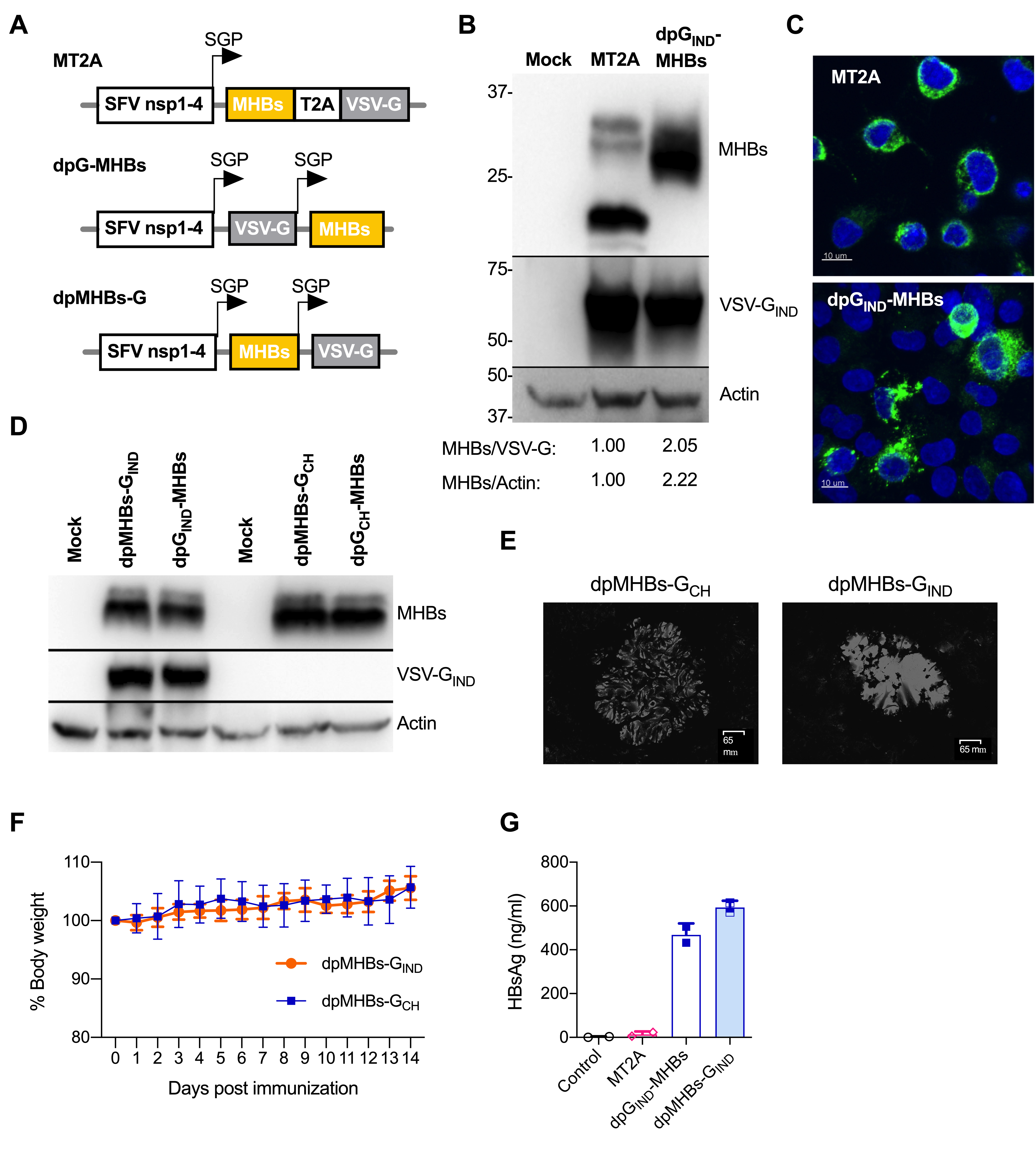
2.1. VLV Dp Vectors
2.2. Peptide Epitopes
2.3. Immunofluorescence
2.4. Immunoblotting
2.5. Fluorescent Anti-HBs Assay
2.6. Immunizations
2.7. AAV-HBV Transduction
2.8. Isolation of Intrahepatic Leukocytes (IHL)
2.9. Intracellular Cytokine Staining by Flow Cytometry
2.10. Enzyme-Linked Immunosorbent Spot (ELISPOT) Assay
2.11. Alanine Aminotransferase (ALT) Measurement
2.12. Enzyme-Linked Immunosorbent Assay (ELISA) Detection of Viral Antigens and Antibodies
2.13. Serum Viral DNA Purification and Detection
2.14. Viral RNA Detection by QPCR
2.15. Statistical Analysis
3. Results
3.1. Improved MHBs Expression and Secretion with Double Subgenomic Promoter VLV
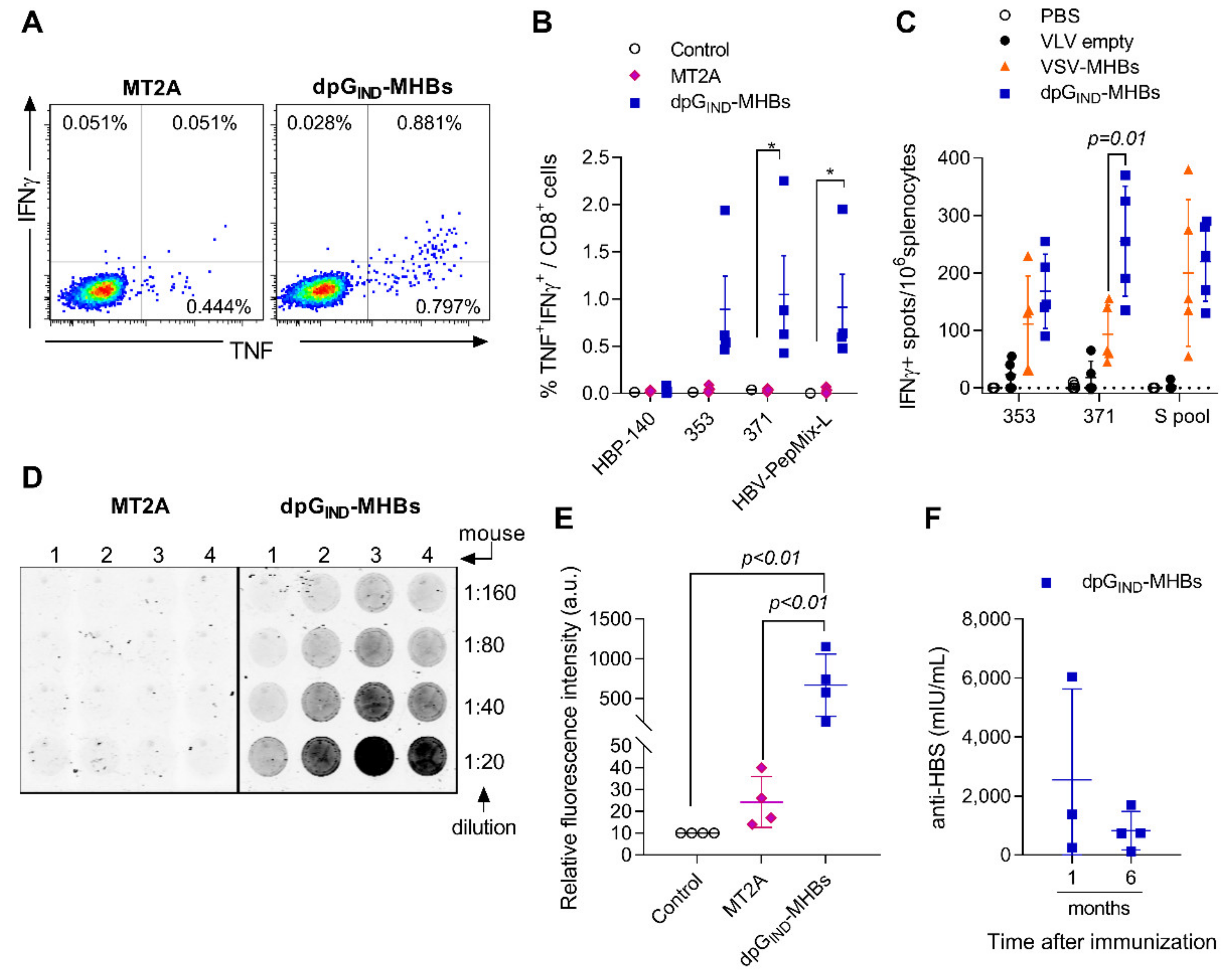
3.2. VLV Dp Generate Enhanced CD8+ T Cell and Antibody Responses
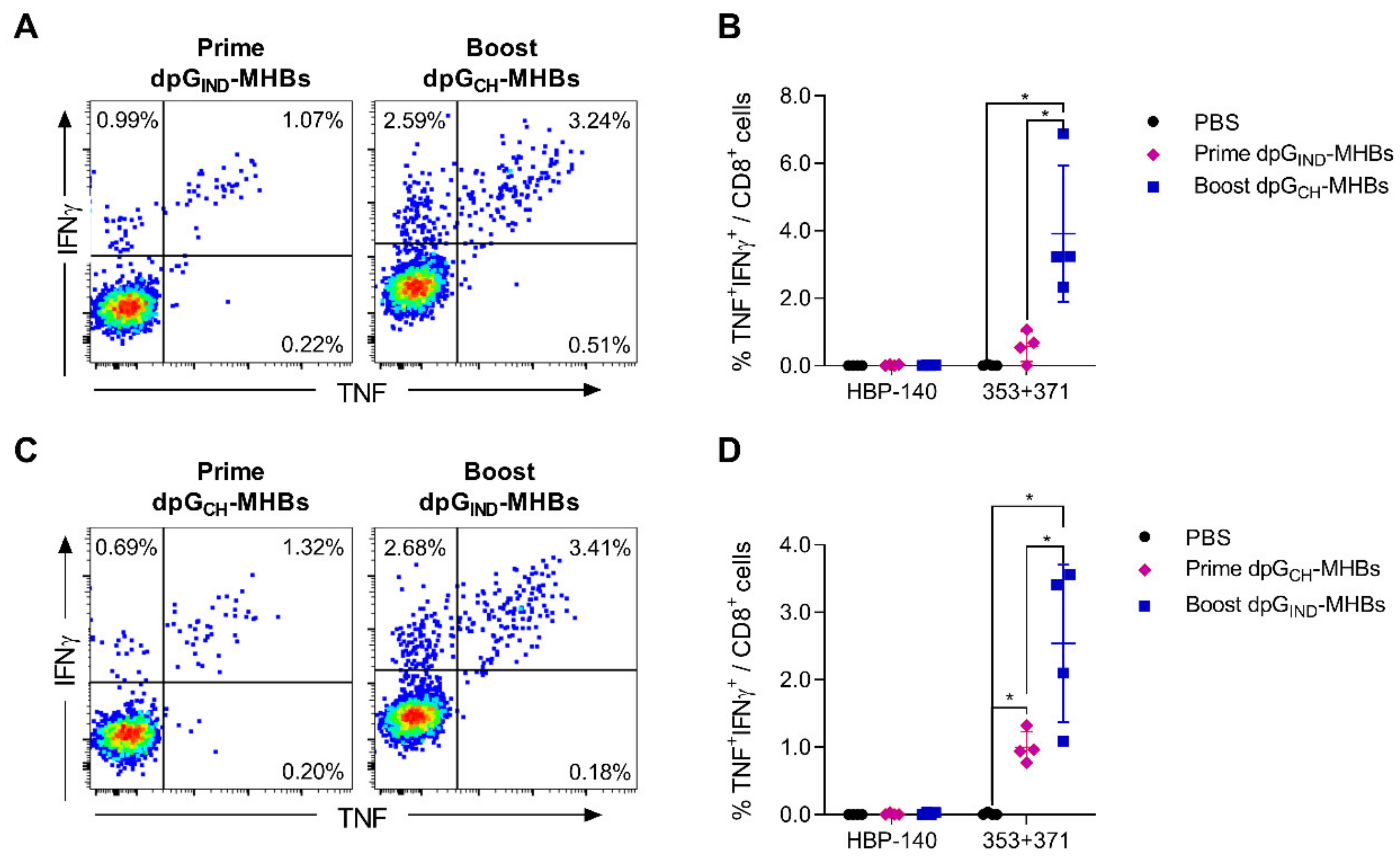
3.3. Homologous Prime-Boost Immunization with Envelope Glycoprotein Switch Enhances the Magnitude of MHBs-Specific CD8+ T Cell Responses
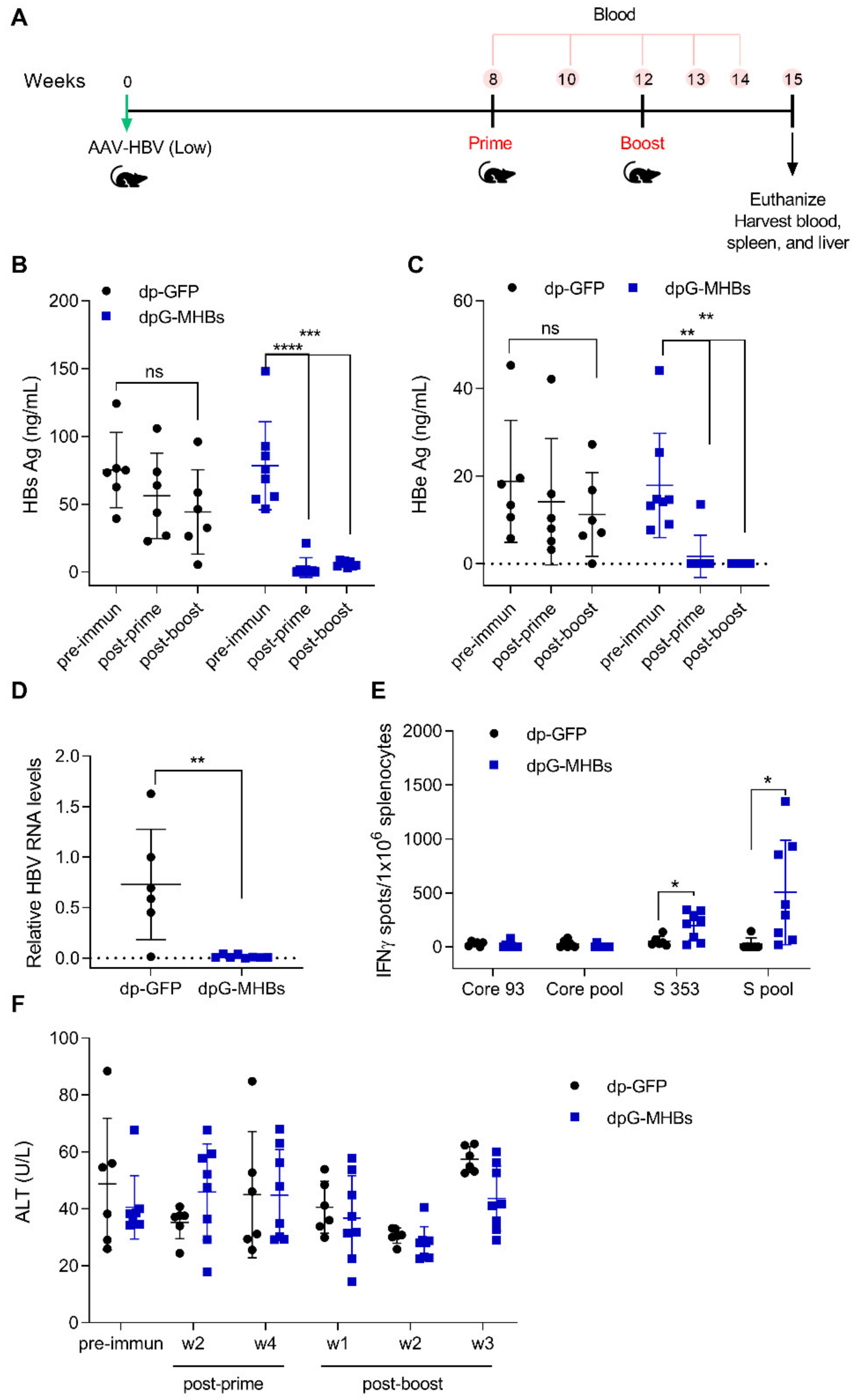
3.4. A Single Dose of VLV Dp Controls Established HBV Replication in Mice with Lower Antigenemia in the AAV-HBV Model
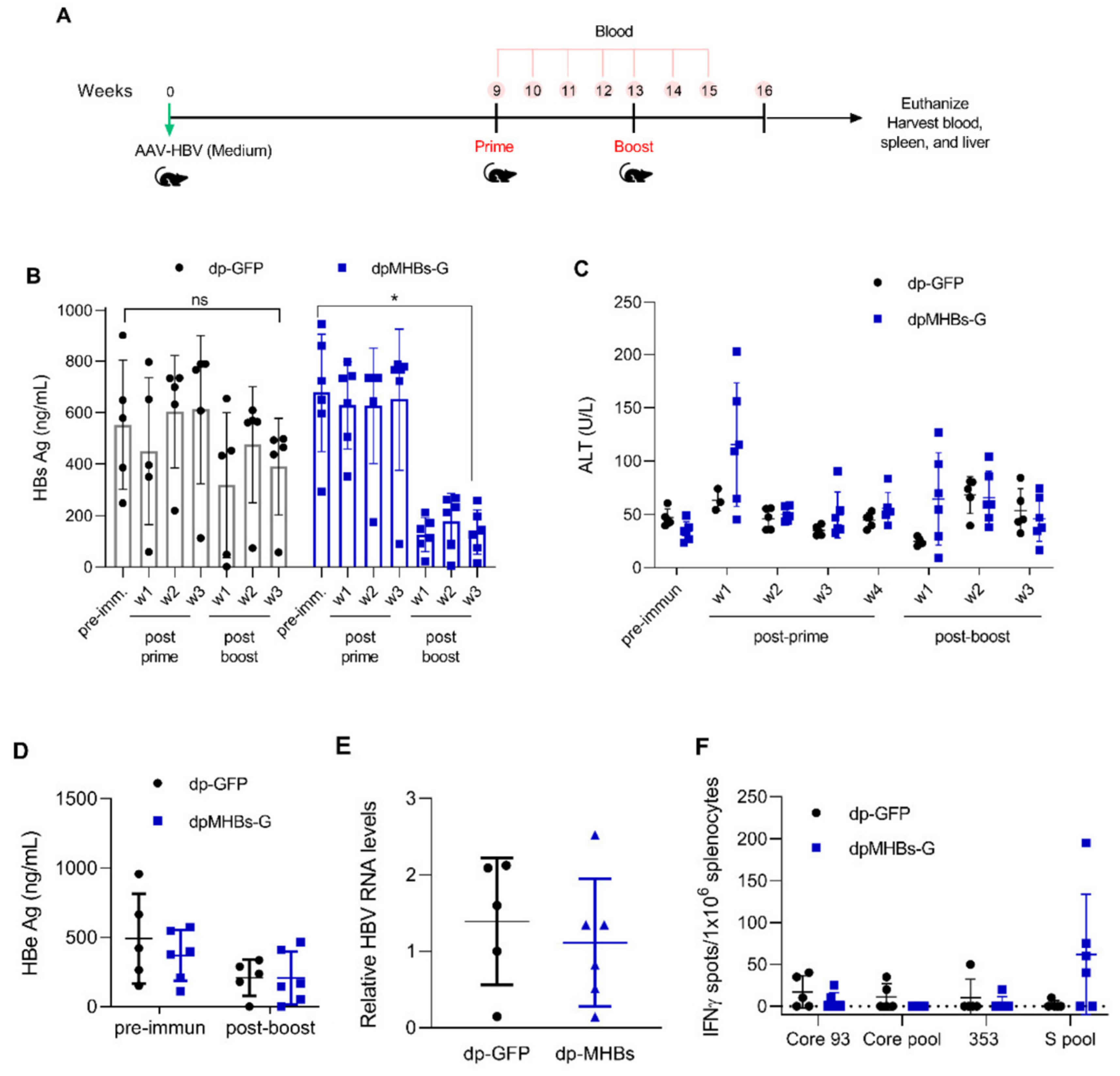
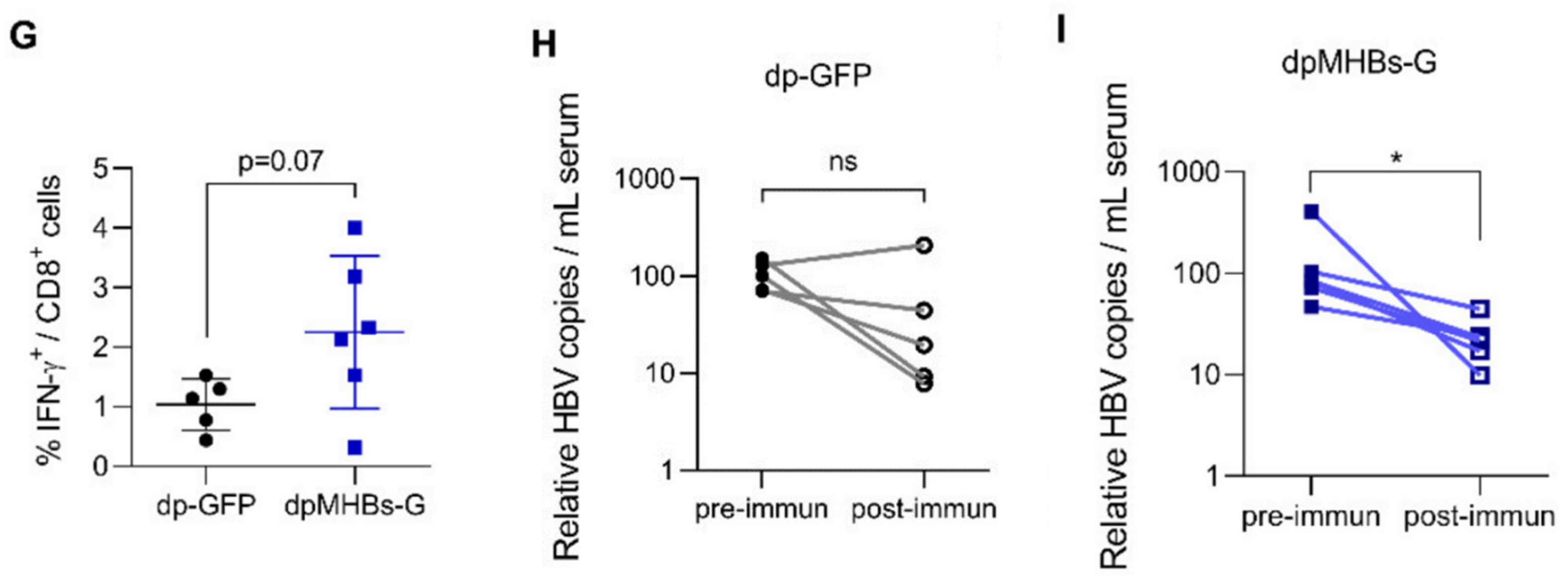
3.5. Prime-Boost with VLV Dp Reduces HBV Levels in Mice with Pre-Existing Intermediate Ag Levels
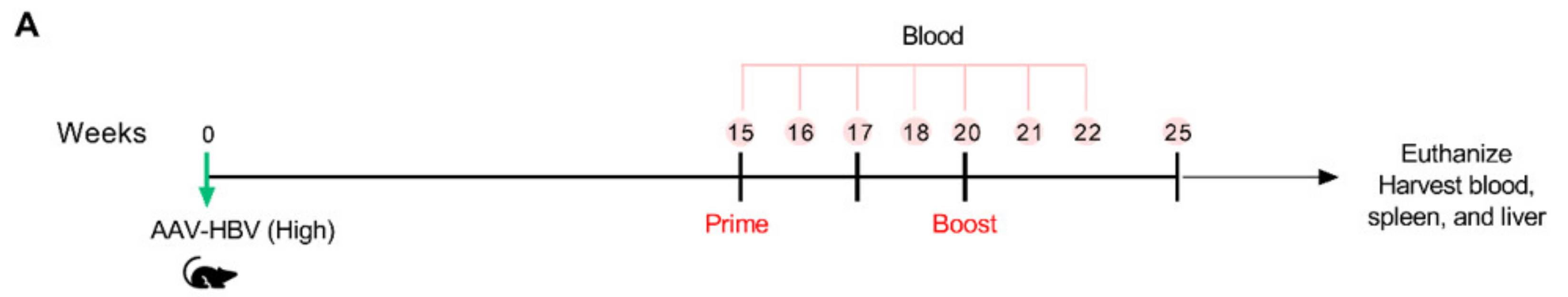
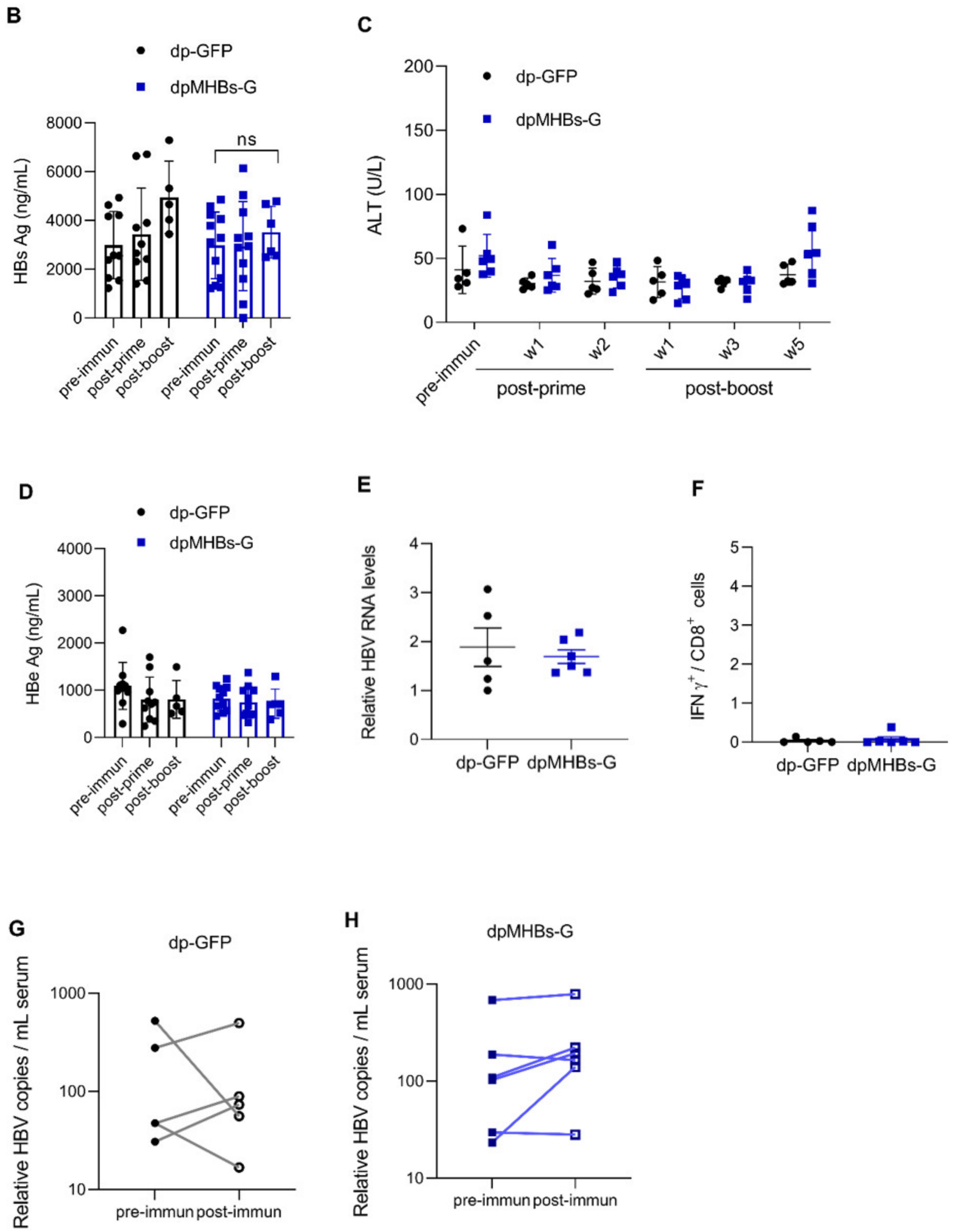
3.6. VLV Dp Prime-Boost Immunization Does Not Control HBV in Mice with Pre-Existing Higher HBV Antigen Levels
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rolls, M.M.; Webster, P.; Balba, N.H.; Rose, J.K. Novel infectious particles generated by expression of the vesicular stomatitis virus glycoprotein from a self-replicating RNA. Cell 1994, 79, 497–506. [Google Scholar] [CrossRef]
- Rolls, M.M.; Haglund, K.; Rose, J.K. Expression of additional genes in a vector derived from a minimal RNA virus. Virology 1996, 218, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.F.; Publicover, J.; Chattopadhyay, A.; Rose, J.K. Hybrid alphavirus-rhabdovirus propagating replicon particles are versatile and potent vaccine vectors. Proc. Natl. Acad. Sci. USA 2008, 105, 5839–5843. [Google Scholar] [CrossRef] [PubMed]
- Schell, J.B.; Rose, N.F.; Bahl, K.; Diller, K.; Buonocore, L.; Hunter, M.; Marx, P.A.; Gambhira, R.; Tang, H.; Montefiori, D.C.; et al. Significant protection against high-dose simian immunodeficiency virus challenge conferred by a new prime-boost vaccine regimen. J. Virol. 2011, 85, 5764–5772. [Google Scholar] [CrossRef] [PubMed]
- Van den Pol, A.N.; Mao, G.; Chattopadhyay, A.; Rose, J.K.; Davis, J.N. Chikungunya, Influenza, Nipah, and Semliki Forest chimeric viruses with vesicular stomatitis virus: Actions in the brain. J. Virol. 2017, 91, e02154-16. [Google Scholar] [CrossRef]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef]
- Rose, N.F.; Buonocore, L.; Schell, J.B.; Chattopadhyay, A.; Bahl, K.; Liu, X.; Rose, J.K. In vitro evolution of high-titer, virus-like vesicles containing a single structural protein. Proc. Natl. Acad. Sci. USA 2014, 111, 16866–16871. [Google Scholar] [CrossRef]
- Reynolds, T.D.; Buonocore, L.; Rose, N.F.; Rose, J.K.; Robek, M.D. Virus-like vesicle-based therapeutic vaccine vectors for chronic hepatitis B virus infection. J. Virol. 2015, 89, 10407–10415. [Google Scholar] [CrossRef]
- Doronina, V.A.; Wu, C.; de Felipe, P.; Sachs, M.S.; Ryan, M.D.; Brown, J.D. Site-specific release of nascent chains from ribosomes at a sense codon. Mol. Cell. Biol. 2008, 28, 4227–4239. [Google Scholar] [CrossRef]
- Yarovinsky, T.O.; Mason, S.W.; Menon, M.; Krady, M.M.; Haslip, M.; Madina, B.R.; Ma, X.; Moshkani, S.; Chiale, C.; Pal, A.C.; et al. Virus-like vesicles expressing multiple antigens for immunotherapy of chronic hepatitis B. iScience 2019, 21, 391–402. [Google Scholar] [CrossRef]
- Hadpech, S.; Jinathep, W.; Saoin, S.; Thongkum, W.; Chupradit, K.; Yasamut, U.; Moonmuang, S.; Tayapiwatana, C. Impairment of a membrane-targeting protein translated from a downstream gene of a “self-cleaving” T2A peptide conjunction. Protein Expr. Purif. 2018, 150, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Dembek, C.; Protzer, U.; Roggendorf, M. Overcoming immune tolerance in chronic hepatitis B by therapeutic vaccination. Curr. Opin. Virol. 2018, 30, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.; Boni, C.; Rossi, M.; Vecchi, A.; Barili, V.; Laccabue, D.; Fisicaro, P.; Missale, G. T cell regulation in HBV-related chronic liver disease. J. Hepatol. 2017, 66, 1096–1098. [Google Scholar] [CrossRef] [PubMed]
- Park, J.J.; Wong, D.K.; Wahed, A.S.; Lee, W.M.; Feld, J.J.; Terrault, N.; Khalili, M.; Sterling, R.K.; Kowdley, K.V.; Bzowej, N.; et al. Hepatitis B virus—Specific and global T-cell dysfunction in chronic hepatitis B. Gastroenterology 2016, 150, 684–695.e5. [Google Scholar] [CrossRef] [PubMed]
- Kosinska, A.D.; Bauer, T.; Protzer, U. Therapeutic vaccination for chronic hepatitis B. Curr. Opin. Virol. 2017, 23, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Couillin, I.; Pol, S.; Mancini, M.; Driss, F.; Brechot, C.; Tiollais, P.; Michel, M.L. Specific vaccine therapy in chronic hepatitis B: Induction of T cell proliferative responses specific for envelope antigens. J. Infect. Dis. 1999, 180, 15–26. [Google Scholar] [CrossRef]
- Pol, S.; Nalpas, B.; Driss, F.; Michel, M.L.; Tiollais, P.; Denis, J.; Brecho, C. Efficacy and limitations of a specific immunotherapy in chronic hepatitis B. J. Hepatol. 2001, 34, 917–921. [Google Scholar] [CrossRef]
- Yalcin, K.; Acar, M.; Degertekin, H. Specific hepatitis B vaccine therapy in inactive HBsAg carriers: A randomized controlled trial. Infection 2003, 31, 221–225. [Google Scholar] [CrossRef]
- Vandepapeliere, P.; Lau, G.K.; Leroux-Roels, G.; Horsmans, Y.; Gane, E.; Tawandee, T.; Merican, M.I.; Win, K.M.; Trepo, C.; Cooksley, G.; et al. Therapeutic vaccination of chronic hepatitis B patients with virus suppression by antiviral therapy: A randomized, controlled study of co-administration of HBsAg/AS02 candidate vaccine and lamivudine. Vaccine 2007, 25, 8585–8597. [Google Scholar] [CrossRef]
- Xu, D.Z.; Wang, X.Y.; Shen, X.L.; Gong, G.Z.; Ren, H.; Guo, L.M.; Sun, A.M.; Xu, M.; Li, L.J.; Guo, X.H.; et al. Results of a phase iii clinical trial with an HBsAg-HBIG immunogenic complex therapeutic vaccine for chronic hepatitis B patients: Experiences and findings. J. Hepatol. 2013, 59, 450–456. [Google Scholar] [CrossRef]
- Fontaine, H.; Kahi, S.; Chazallon, C.; Bourgine, M.; Varaut, A.; Buffet, C.; Godon, O.; Meritet, J.F.; Saidi, Y.; Michel, M.L.; et al. Anti- DNA vaccination does not prevent relapse after discontinuation of analogues in the treatment of chronic hepatitis B: A randomised trial--ANRS HB02 VAC-ADN. Gut 2015, 64, 139–147. [Google Scholar] [CrossRef]
- Godon, O.; Fontaine, H.; Kahi, S.; Meritet, J.F.; Scott-Algara, D.; Pol, S.; Michel, M.L.; Bourgine, M.; ANRS HB02 Study Group. Immunological and antiviral responses after therapeutic DNA immunization in chronic hepatitis B patients efficiently treated by analogues. Mol. Ther. 2014, 22, 675–684. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ewer, K.J.; Lambe, T.; Rollier, C.S.; Spencer, A.J.; Hill, A.V.; Dorrell, L. Viral vectors as vaccine platforms: From immunogenicity to impact. Curr. Opin. Immunol. 2016, 41, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E. Therapeutic vaccines in HBV: Lessons from HCV. Med. Microbiol. Immunol. 2015, 204, 79–86. [Google Scholar] [CrossRef][Green Version]
- Kutscher, S.; Bauer, T.; Dembek, C.; Sprinzl, M.; Protzer, U. Design of therapeutic vaccines: Hepatitis B as an example. Microb. Biotechnol. 2012, 5, 270–282. [Google Scholar] [CrossRef]
- Chiale, C.; Moshkani, S.; Rose, J.K.; Robek, M.D. Heterologous prime-boost immunization with vesiculovirus-based vectors expressing HBV core antigen induces CD8(+) T cell responses in naive and persistently infected mice and protects from challenge. Antiviral Res. 2019, 168, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.F.; Roberts, A.; Buonocore, L.; Rose, J.K. Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J. Virol. 2000, 74, 10903–10910. [Google Scholar] [CrossRef]
- Cobleigh, M.A.; Buonocore, L.; Uprichard, S.L.; Rose, J.K.; Robek, M.D. A vesicular stomatitis virus-based hepatitis B virus vaccine vector provides protection against challenge in a single dose. J. Virol. 2010, 84, 7513–7522. [Google Scholar] [CrossRef]
- Cobleigh, M.A.; Wei, X.; Robek, M.D. A vesicular stomatitis virus-based therapeutic vaccine generates a functional CD8 T cell response to hepatitis B virus in transgenic mice. J. Virol. 2013, 87, 2969–2973. [Google Scholar] [CrossRef]
- Schirmbeck, R.; Bohm, W.; Fissolo, N.; Melber, K.; Reimann, J. Different immunogenicity of h-2 kb-restricted epitopes in natural variants of the hepatitis B surface antigen. Eur. J. Immunol. 2003, 33, 2429–2438. [Google Scholar] [CrossRef]
- Sette, A.D.; Oseroff, C.; Sidney, J.; Alexander, J.; Chesnut, R.W.; Kakimi, K.; Guidotti, L.G.; Chisari, F.V. Overcoming t cell tolerance to the hepatitis B virus surface antigen in hepatitis B virus-transgenic mice. J. Immunol. 2001, 166, 1389–1397. [Google Scholar] [CrossRef]
- Reynolds, T.D.; Moshkani, S.; Robek, M.D. An elispot-based assay to measure HBV-specific CD(+) T cell responses in immunocompetent mice. Methods Mol. Biol. 2017, 1540, 237–247. [Google Scholar]
- Garson, J.A.; Grant, P.R.; Ayliffe, U.; Ferns, R.B.; Tedder, R.S. Real-time PCR quantitation of hepatitis B virus DNA using automated sample preparation and murine cytomegalovirus internal control. J. Virol. Methods 2005, 126, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mehta, A.; Dwek, R.; Butters, T.; Block, T. Evidence that N-linked glycosylation is necessary for hepatitis B virus secretion. Virology 1995, 213, 660–665. [Google Scholar] [CrossRef]
- Lefrancois, L. Protection against lethal viral infection by neutralizing and nonneutralizing monoclonal antibodies: Distinct mechanisms of action in vivo. J.Virol. 1984, 51, 208–214. [Google Scholar] [CrossRef]
- Steinhoff, U.; Muller, U.; Schertler, A.; Hengartner, H.; Aguet, M.; Zinkernagel, R.M. Antiviral protection by vesicular stomatitis virus-specific antibodies in alpha/beta interferon receptor-deficient mice. J. Virol. 1995, 69, 2153–2158. [Google Scholar] [CrossRef]
- Bengsch, B.; Martin, B.; Thimme, R. Restoration of HBV-specific CD8+ T cell function by pd-1 blockade in inactive carrier patients is linked to t cell differentiation. J. Hepatol. 2014, 61, 1212–1219. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Boni, C.; Barili, V.; Laccabue, D.; Ferrari, C. Strategies to overcome HBV-specific T cell exhaustion: Checkpoint inhibitors and metabolic re-programming. Curr. Opin. Virol. 2018, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Ravanetti, L.; Urbani, S.; Giuberti, T.; Cavalli, A.; Vandelli, C.; Andreone, P.; et al. Combined blockade of programmed death-1 and activation of CD137 increase responses of human liver t cells against HBV, but not HCV. Gastroenterology 2012, 143, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Jacobi, F.J.; Wild, K.; Smits, M.; Zoldan, K.; Csernalabics, B.; Flecken, T.; Lang, J.; Ehrenmann, P.; Emmerich, F.; Hofmann, M.; et al. OX40 stimulation and pd-l1 blockade synergistically augment HBV-specific CD4 t cells in patients with HBeAg-negative infection. J. Hepatol. 2019, 70, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Publicover, J.; Gaggar, A.; Jespersen, J.M.; Halac, U.; Johnson, A.J.; Goodsell, A.; Avanesyan, L.; Nishimura, S.L.; Holdorf, M.; Mansfield, K.G.; et al. An OX40/OX40L interaction directs successful immunity to hepatitis B virus. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar] [PubMed]
- Masters, P.S.; Bhella, R.S.; Butcher, M.; Patel, B.; Ghosh, H.P.; Banerjee, A.K. Structure and expression of the glycoprotein gene of Chandipura virus. Virology 1989, 171, 285–290. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiale, C.; Yarovinsky, T.O.; Mason, S.W.; Madina, B.R.; Menon, M.; Krady, M.M.; Moshkani, S.; Chattopadhyay Pal, A.; Almassian, B.; Rose, J.K.; et al. Modified Alphavirus-Vesiculovirus Hybrid Vaccine Vectors for Homologous Prime-Boost Immunotherapy of Chronic Hepatitis B. Vaccines 2020, 8, 279. https://doi.org/10.3390/vaccines8020279
Chiale C, Yarovinsky TO, Mason SW, Madina BR, Menon M, Krady MM, Moshkani S, Chattopadhyay Pal A, Almassian B, Rose JK, et al. Modified Alphavirus-Vesiculovirus Hybrid Vaccine Vectors for Homologous Prime-Boost Immunotherapy of Chronic Hepatitis B. Vaccines. 2020; 8(2):279. https://doi.org/10.3390/vaccines8020279
Chicago/Turabian StyleChiale, Carolina, Timur O. Yarovinsky, Stephen W. Mason, Bhaskara R. Madina, Manisha Menon, Marie M. Krady, Safiehkhatoon Moshkani, Anasuya Chattopadhyay Pal, Bijan Almassian, John K. Rose, and et al. 2020. "Modified Alphavirus-Vesiculovirus Hybrid Vaccine Vectors for Homologous Prime-Boost Immunotherapy of Chronic Hepatitis B" Vaccines 8, no. 2: 279. https://doi.org/10.3390/vaccines8020279
APA StyleChiale, C., Yarovinsky, T. O., Mason, S. W., Madina, B. R., Menon, M., Krady, M. M., Moshkani, S., Chattopadhyay Pal, A., Almassian, B., Rose, J. K., Robek, M. D., & Nakaar, V. (2020). Modified Alphavirus-Vesiculovirus Hybrid Vaccine Vectors for Homologous Prime-Boost Immunotherapy of Chronic Hepatitis B. Vaccines, 8(2), 279. https://doi.org/10.3390/vaccines8020279