Porcine Parvovirus 7: Evolutionary Dynamics and Identification of Epitopes toward Vaccine Design


Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Datasets
2.2. Multiple Sequence Alignment
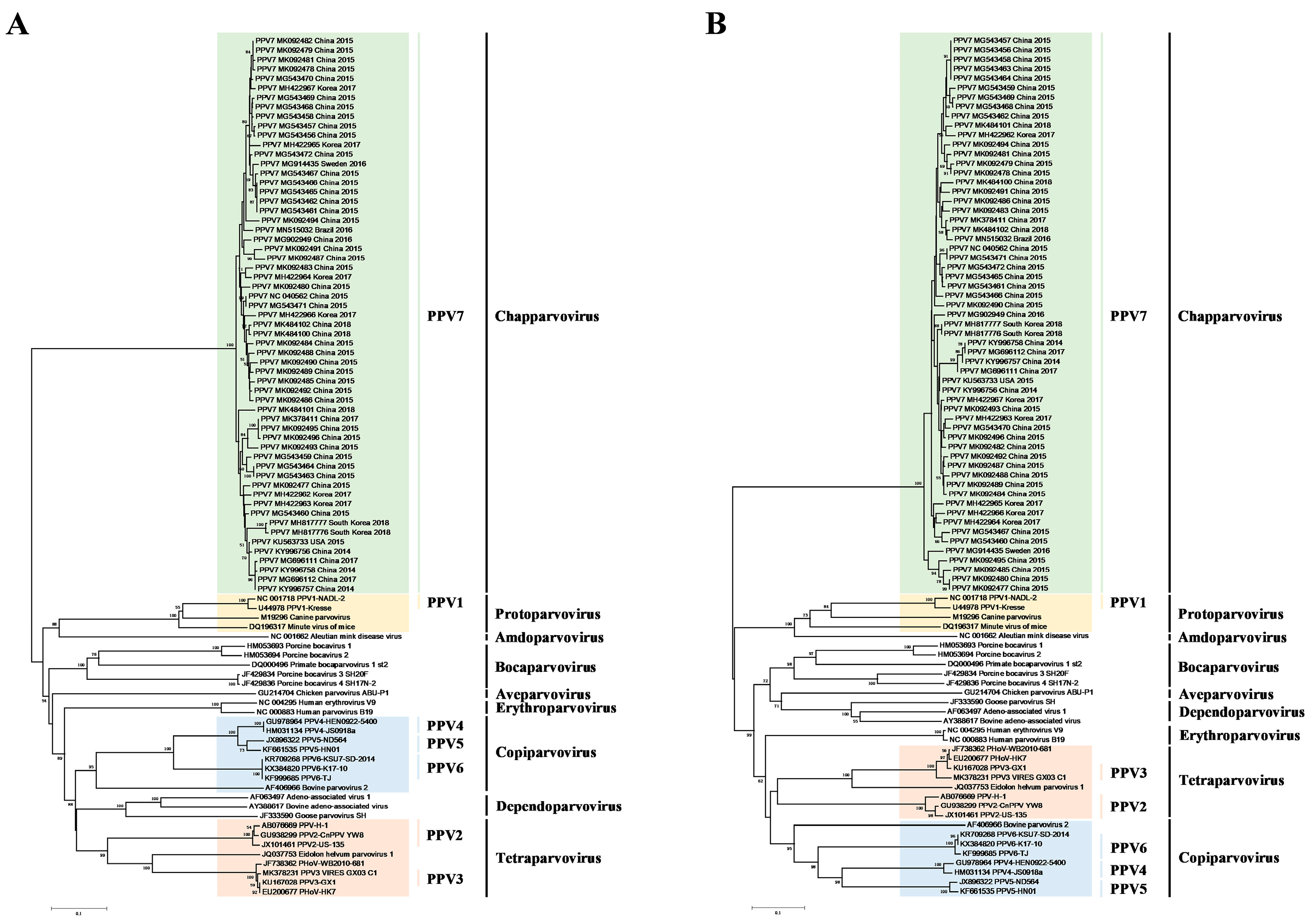
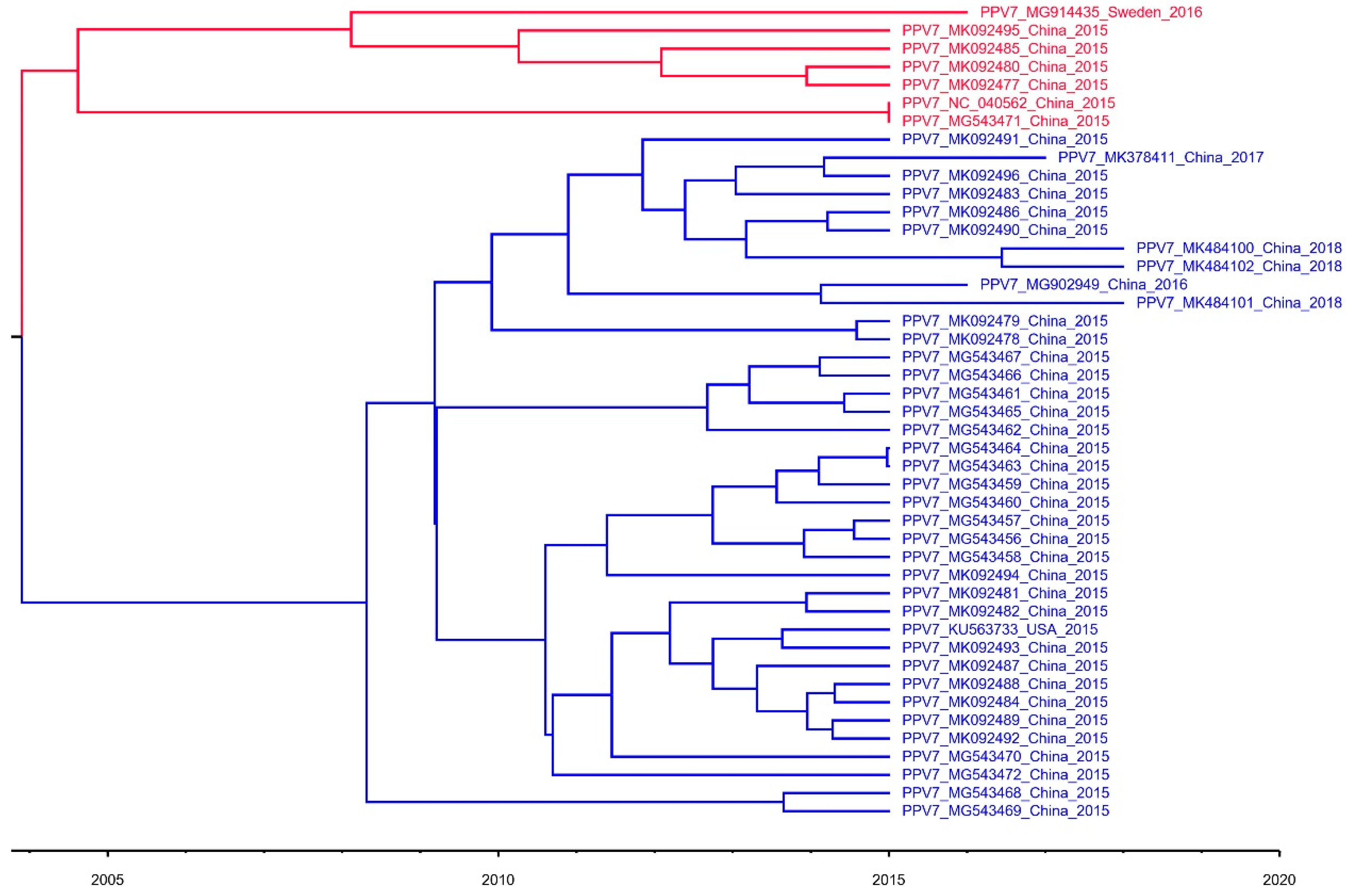
2.3. Phylogenetic and Evolution Dynamic Analysis
2.4. Selection Pressures Analysis
2.5. Structural Analysis
2.6. B Cell Epitope Prediction
2.7. CD8 T Cell Epitope Prediction
2.8. Peptide Modelling and Molecular Docking
3. Results
3.1. Construction of Phylogenetic Tree and Evolution Dynamic Analysis
3.2. Selection Pressures Analysis
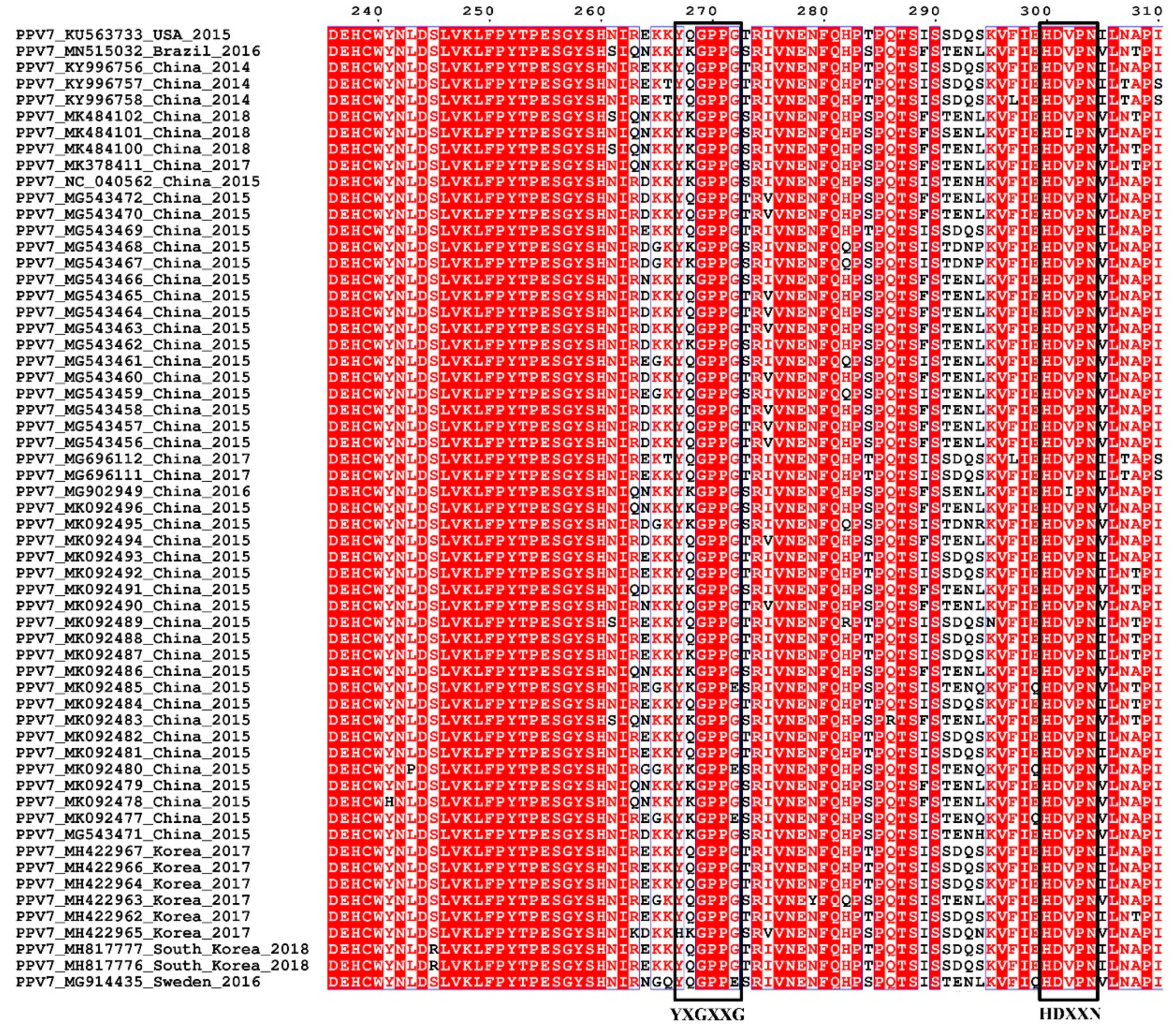
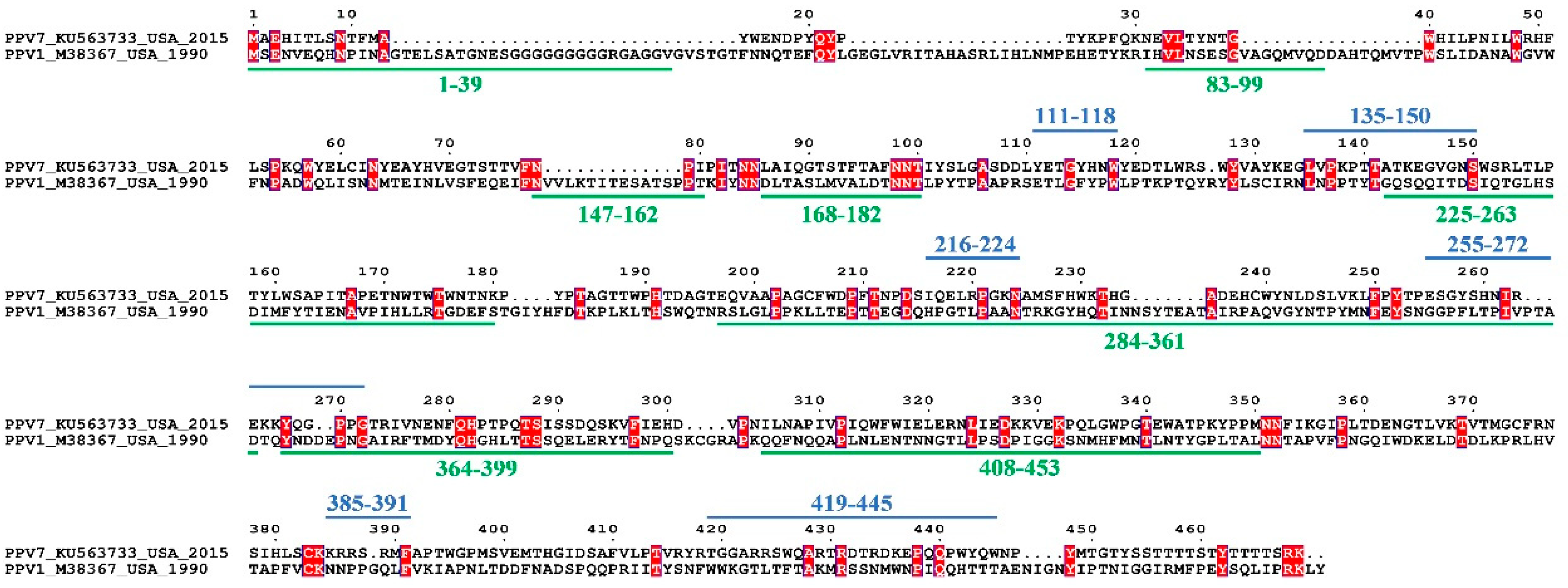
3.3. Sequence and Structural Characteristics
3.4. Linear B Cell Epitopes Prediction and Analysis of Cap
3.5. CD8 T Cell Epitopes Prediction and Interaction Study of Predicted Peptides with SLA Alleles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-Mckenna, M.; Chiorini, J.A.; Mukha, D.V.; Davison, A. The family Parvoviridae. Arch. Virol. 2013, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Palinski, R.M.; Mitra, N.; Hause, B.M. Discovery of a novel Parvovirinae virus, porcine parvovirus 7, by metagenomic sequencing of porcine rectal swabs. Virus Genes 2016, 52, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Canal, C.W.; Truyen, U. Molecular epidemiology and evolution of porcine parvoviruses. Infect. Genet. Evol. 2015, 36, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Mahnel, H. Cultivation of hog cholera virus in pig kidney cultures with cytopathogenic effect. Zent. Bakteriol Orig 1965, 195, 157–166. [Google Scholar]
- Mengeling, W.L.; Lager, K.M.; Vorwald, A.C. The effect of porcine parvovirus and porcine reproductive and respiratory syndrome virus on porcine reproductive performance. Anim. Reprod. Sci. 2000, 60, 199–210. [Google Scholar] [CrossRef]
- Hueffer, K.; Parrish, C. Parvovirus host range, cell tropism and evolution. Curr. Opin. Microbiol. 2003, 6, 392–398. [Google Scholar] [CrossRef]
- Hijikata, M.; Abe, K.; Win, K.M.; Shimizu, Y.K.; Yoshikura, H. Identification of new parvovirus DNA sequence in swine sera from Myanmar. Jpn. J. Infect. Dis. 2002, 54, 244–245. [Google Scholar]
- Lau, S.K.P.; Woo, P.C.Y.; Tse, H.; Fu, C.T.Y.; Au, W.-K.; Chen, X.-C.; Tsoi, H.-W.; Tsang, T.H.F.; Chan, J.S.Y.; Tsang, D.N.C.; et al. Identification of novel porcine and bovine parvoviruses closely related to human parvovirus. J. Gen. Virol. 2008, 89, 1840–1848. [Google Scholar] [CrossRef]
- Cheung, A.K.; Wu, G.; Wang, D.; Bayles, D.O.; Lager, K.M.; Vincent, A.L. Identification and molecular cloning of a novel porcine parvovirus. Arch. Virol. 2010, 155, 801–806. [Google Scholar] [CrossRef]
- Xiao, C.T.; Giménez-Lirola, L.G.; Jiang, Y.-H.; Halbur, P.G.; Opriessnig, T.; Elankumaran, S. Characterization of a novel porcine parvovirus tentatively designated PPV5. PLoS ONE 2013, 8, e65312. [Google Scholar] [CrossRef]
- Ni, J.; Qiao, C.; Han, X.; Han, T.; Kang, W.; Zi, Z.; Cao, Z.; Zhai, X.; Cai, X. Identification and genomic characterization of a novel porcine parvovirus (PPV6) in China. Virol. J. 2014, 11, 203. [Google Scholar] [CrossRef]
- Anne-Lie, B.M.; Ye, X.; Caroline, F.; Per, W.; Mikael, B. Characterisation of the virome of tonsils from conventional pigs and from specific pathogen-free pigs. Viruses 2018, 10, 382. [Google Scholar]
- Miłek, D.; Woźniak, A.; Stadejek, T. The detection and genetic diversity of novel porcine parvovirus 7 (PPV7) on Polish pig farms. Res. Vet. Sci. 2018, 120, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Zhou, H.; Tong, L.; Chen, Y.; Sun, Y.; Wang, H.; Zhang, G. First identification of porcine parvovirus 7 in China. Arch. Virol. 2018, 163, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, K.-K.; Wang, J.; Wang, X.-P.; Zhao, L.; Sun, P.; Li, Y.-D. Detection and molecular characterization of novel porcine parvovirus 7 in Anhui province from Central-Eastern China. Infect. Genet. Evol. 2019, 71, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cao, L.; Sun, W.; Xin, J.; Zheng, M.; Tian, M.; Lu, H.; Jin, N. Sequence and phylogenetic analysis of novel porcine parvovirus 7 isolates from pigs in Guangxi, China. PLoS ONE 2019, 14, e0219560. [Google Scholar] [CrossRef]
- Ouh, I.-O.; Park, S.; Lee, J.-Y.; Song, J.Y.; Cho, I.-S.; Kim, H.-R.; Park, C.-K. First detection and genetic characterization of porcine parvovirus 7 from Korean domestic pig farms. J. Vet. Sci. 2018, 19, 855–857. [Google Scholar] [CrossRef]
- Da Silva, M.S.; Budaszewski, R.F.; Weber, M.N.; Cibulski, S.P.; Paim, W.P.; Mósena, A.C.S.; Canova, R.; Varela, A.P.M.; Mayer, F.Q.; Pereira, C.W.; et al. Liver virome of healthy pigs reveals diverse small ssDNA viral genomes. Infect. Genet. Evol. 2020, 81, 104203. [Google Scholar] [CrossRef]
- Chung, H.-C.; Nguyen, V.-G.; Huynh, T.-M.-L.; Park, Y.-H.; Park, K.-T.; Park, B.-K. PCR-based detection and genetic characterization of porcine parvoviruses in South Korea in 2018. BMC Vet. Res. 2020, 16, 113. [Google Scholar] [CrossRef]
- Antonis, A.F.G.; Bruschke, C.J.M.; Rueda, P.; Maranga, L.; Casal, J.I.; Vela, C.; Hilgers, L.A.T.; Belt, P.B.G.M.; Weerdmeester, K.; Carrondo, M.J.T. A novel recombinant virus-like particle vaccine for prevention of porcine parvovirus-induced reproductive failure. Vaccine 2006, 24, 5481–5490. [Google Scholar] [CrossRef]
- Ji, P.; Liu, Y.; Chen, Y.; Wang, A.; Jiang, D.; Zhao, B.; Wang, J.; Chai, S.; Zhou, E.; Zhang, G. Porcine parvovirus capsid protein expressed in Escherichia coli self-assembles into virus-like particles with high immunogenicity in mice and guinea pigs. Antivir. Res. 2017, 139, 146–152. [Google Scholar] [CrossRef]
- Saade, G.; Deblanc, C.; Bougon, J.; Marois-Créhan, C.; Fablet, C.; Auray, G.; Belloc, C.; Leblanc-Maridor, M.; Gagnon, C.A.; Zhu, J.; et al. Coinfections and their molecular consequences in the porcine respiratory tract. Vet. Res. 2020, 51, 80. [Google Scholar] [CrossRef] [PubMed]
- Miłek, D.; Woźniak, A.; Podgórska, K.; Stadejek, T. Do porcine parvoviruses 1 through 7 (PPV1-PPV7) have an impact on porcine circovirus type 2 (PCV2) viremia in pigs? Vet. Microbiol. 2020, 242, 108613. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky, P.S.L.; Frost, S.D.W. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Ben, M.; Sasha, M.; Amandla, M.; Thomas, W.; Daniel, S.; Pond, S.L.K.; Konrad, S. FUBAR: A fast, unconstrained bayesian AppRoximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Hughes, A.L.; Nei, M. Nucleotide substitution at major histocompatibility complex class II loci: Evidence for overdominant selection. Proc. Natl. Acad. Sci. USA 1989, 86, 958–962. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Minneci, F.; Nugent, T.; Bryson, K.; Jones, D.T. Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013, 41, W349–W357. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.E.; Harndahl, M.; Nielsen, M.; Patch, J.R.; Jungersen, G.; Buus, S.; Golde, W.T. Identification of peptides from foot-and-mouth disease virus structural proteins bound by class I swine leukocyte antigen (SLA) alleles, SLA-1*0401 and SLA-2*0401. Anim. Genet. 2013, 44, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.E.; Rasmussen, M.; Harndahl, M.; Nielsen, M.; Buus, S.; Jungersen, G. A combined prediction strategy increases identification of peptides bound with high affinity and stability to porcine MHC class I molecules SLA-1*04:01, SLA-2*04:01, and SLA-3*04:01. Immunogenetics 2016, 68, 157–165. [Google Scholar] [CrossRef]
- Gutiérrez, A.H.; Martin, W.D.; Bailey-Kellogg, C.; Terry, F.; Moise, L.; Groot, A.S.D. Development and validation of an epitope prediction tool for swine (PigMatrix) based on the pocket profile method. BMC Bioinform. 2015, 16, 290. [Google Scholar] [CrossRef] [PubMed]
- Alexis, L.; Pierre, T.; Julien, R.; Marek, V.; Philippe, D.; Pierre, T.J.N.A.R. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: Fast interaction refinement in molecular docking. Proteins Struct. Funct. Bioinform. 2007, 69, 139–159. [Google Scholar] [CrossRef]
- Efrat, M.; Dina, S.D.; Nelly, A.; Ruth, N.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, W229–W232. [Google Scholar]
- Ren, X.; Tao, Y.; Cui, J.; Suo, S.; Cong, Y.; Tijssen, P. Phylogeny and evolution of porcine parvovirus. Virus Res. 2013, 178, 392–397. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef]
- Cadar, D.; Lo˝rincz, M.; Kiss, T.; Novosel, D.; Podgorska, K.; Becskei, Z.; Tuboly, T.s.; Csa´gola, A. Emerging novel porcine parvoviruses in Europe: Origin, evolution, phylodynamics and phylogeography. J. Gen. Virol. 2013, 94, 2330–2337. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Bonatto, S.L.; Homeier, T.; Souza, C.K.; Truyen, U. High rate of viral evolution in the capsid protein of porcine parvovirus. J. Gen. Virol. 2011, 92, 2628–2636. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Söderlund-Venermo, M.; Young, N.S. Human parvoviruses. Clin. Microbiol. Rev. 2017, 30, 43–113. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chatterjee, K.; Chattopadhyay, N.R.; Choudhuri, T. Evolutionary aspects of Parvovirus B-19V associated diseases and their pathogenesis patterns with an emphasis on vaccine development. Virusdisease 2019, 30, 32–42. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FEL | SLAC | FUBAR | MEME | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Site | dN-dS | p-Value | Site | dN-dS | p-Value | Site | dN-dS | Post.Pro | Site | β+ | p-Value |
| 24 | 2.492 | 0.004 | 24 | 5.741 | 0.018 | 24 | 6.900 | 0.997 | 24 | 2.5 | 0.01 |
| 106 | 2.157 | 0.023 | 106 | 4.943 | 0.028 | 106 | 5.706 | 0.979 | 106 | 24.21 | 0 |
| 158 | 1.552 | 0.033 | 158 | 3.652 | 0.098 | 158 | 3.244 | 0.972 | 158 | 1.55 | 0.05 |
| 195 | 1.442 | 0.909 | 195 | 166.2 | 0 | ||||||
| 270 | 1.777 | 0.026 | 270 | 5.107 | 0.048 | 270 | 4.363 | 0.974 | 270 | 14.87 | 0 |
| 441 | 2.926 | 0.944 | 441 | 18.72 | 0 | ||||||
| 446 | 1.649 | 0.015 | 446 | 3.970 | 0.055 | 446 | 3.901 | 0.988 | 446 | 3.43 | 0.02 |
| FEL | SLAC | FUBAR | MEME | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Site | dN-dS | p-Value | Site | dN-dS | p-Value | Site | dN-dS | Post.Pro | Site | β+ | p-Value |
| 27 | 1.519 | 0.016 | 27 | 2.24 | 0.977 | 27 | 1.52 | 0.03 | |||
| 69 | 2.815 | 0.002 | 69 | 3.466 | 0.069 | 69 | 4.953 | 0.997 | 69 | 2.82 | 0 |
| 83 | 2.627 | 0.96 | 83 | 148.66 | 0 | ||||||
| 342 | 5.477 | 0.01 | 342 | 8.331 | 0.005 | 342 | 12.563 | 0.993 | 342 | 173.79 | 0 |
| 376 | 4.566 | 0.066 | 376 | 5.16 | 0.965 | ||||||
| 426 | 8.215 | 0.005 | 426 | 10.973 | 0.002 | 426 | 17.289 | 0.999 | 426 | 40.1 | 0 |
| 465 | 1.905 | 0.034 | 465 | 3.658 | 0.088 | 465 | 2.882 | 0.979 | 465 | 1.9 | 0.05 |
| 474 | 4.118 | 0.019 | 474 | 5.486 | 0.026 | 474 | 7.406 | 0.983 | 474 | 4.12 | 0.03 |
| 498 | 4.261 | 0 | 498 | 5.326 | 0.031 | 498 | 8.12 | 1 | 498 | 4.25 | 0 |
| 500 | 5.817 | 0.028 | 500 | 2.963 | 0.956 | ||||||
| 525 | 7.599 | 0.003 | 525 | 10.047 | 0.002 | 525 | 15.702 | 0.999 | 525 | 7.6 | 0 |
| 547 | 5.486 | 0.026 | 547 | 4.412 | 0.97 | ||||||
| 549 | 4.267 | 0.059 | 549 | 2.533 | 0.956 | ||||||
| 550 | 3.658 | 0.088 | 550 | 2.944 | 0.992 | 550 | 1.9 | 0.01 | |||
| 579 | 2.081 | 0.01 | 579 | 3.455 | 0.994 | 579 | 2.08 | 0.02 | |||
| 603 | 1.271 | 0.025 | 603 | 3.778 | 0.081 | 603 | 1.998 | 0.974 | 603 | 1.27 | 0.04 |
| 618 | 4.105 | 0.077 | 618 | 2.663 | 0.95 | ||||||
| Epitope | Start | End | Sequence | Length | Identity (59 Isolates) | Vaxijen v2.0 Score |
|---|---|---|---|---|---|---|
| A | 111 | 118 | YETGYHNW | 8 | 72.88% | 0.5888 |
| B | 135 | 150 | LVPKPTTATKEGVGNS | 16 | 18.64% | 0.4355 |
| C | 216 | 224 | IQELRPGKN | 9 | 98.31% | 0.7908 |
| D | 255 | 272 | ESGYSHNIREKKYQGPPG | 18 | 25.42% | 0.6235 |
| E | 385 | 391 | KRRSRMF | 7 | 100.00% | 1.6044 |
| F | 419 | 445 | TGGARRSWQARTRDTRDKEPQQPWYQW | 27 | 10.17% | 0.4966 |
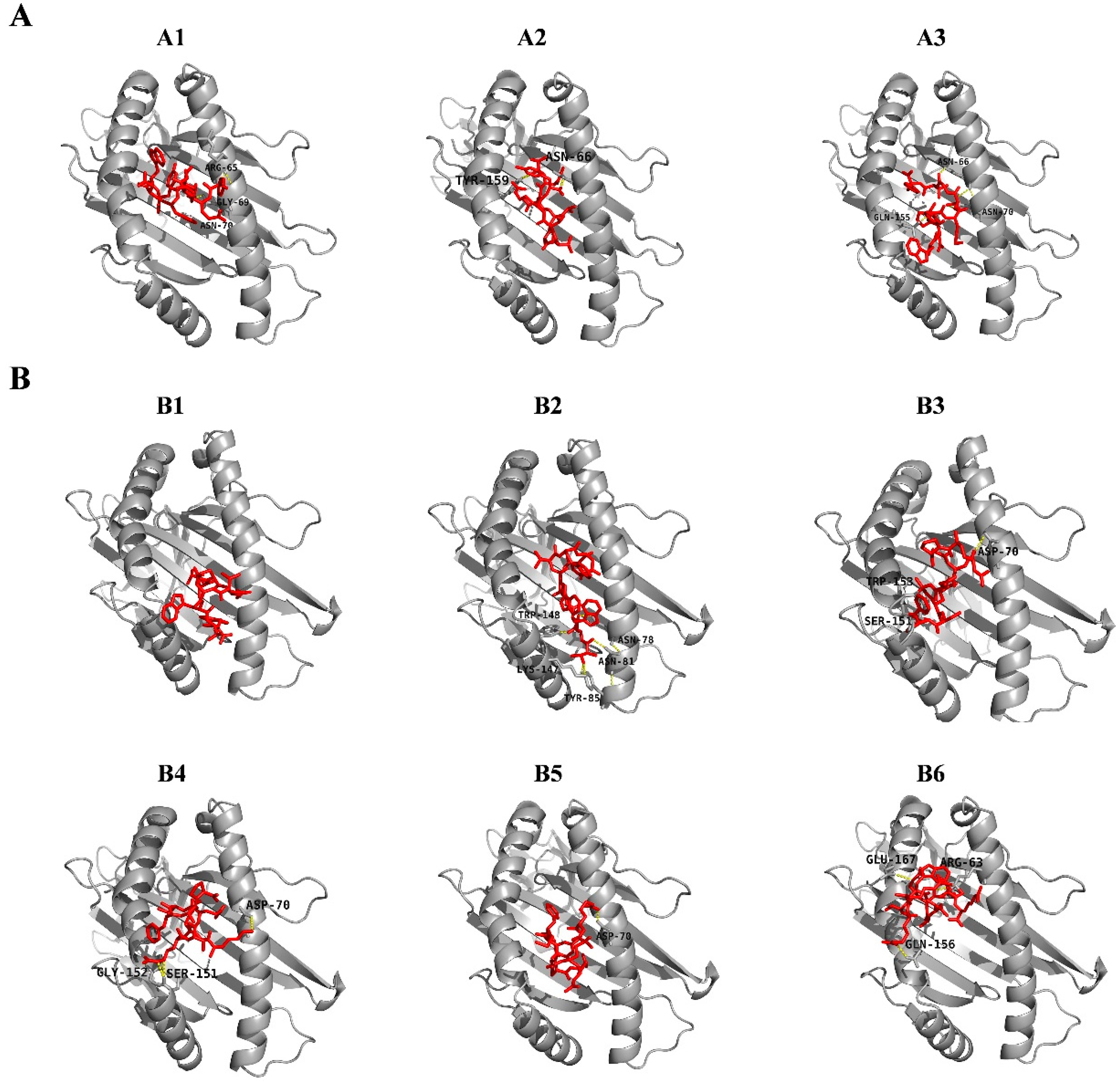
| No. | Allele | Start | End | Peptide | Global Energy (kcal/mol) | vdW Energy (kcal/mol) | H-Bond Energy (kcal/mol) | Interacting Residues | Vaxijen Score | Identity (59 Isolates) |
|---|---|---|---|---|---|---|---|---|---|---|
| A1 | SLA-2*0402 | 111 | 119 | YETGYHNWY | −26.32 | −22.21 | −1.43 | Arg65, Gly69, Asn70 | 0.5659 | 72.88% |
| A2 | SLA-2*0402 | 409 | 417 | AFVLPTVRY | −23.20 | −24.38 | −1.49 | Asn66, Tyr159 | 0.7382 | 91.53% |
| A3 | SLA-2*0402 | 445 | 453 | WNPYMTGTY | −54.08 | −30.77 | −3.19 | Asn66, Asn70, Gln155 | 1.2177 | 32.20% |
| B1 | SLA-3*0401 | 166 | 174 | TAPETNWTW | −25.49 | −21.17 | −0.82 | 1.4351 | 96.61% | |
| B2 | SLA-3*0401 | 170 | 178 | TNWTWTWNT | −37.42 | −22.35 | −4.1 | Asn78, Asn81, Tyr85, Lys147, Trp148 | 1.2242 | 33.90% |
| B3 | SLA-3*0401 | 220 | 228 | RPGKNAMSF | −44.82 | −24.06 | −1.49 | Asp70, Ser151, Trp153 | 0.5339 | 91.53% |
| B4 | SLA-3*0401 | 385 | 393 | KRRSRMFAP | −33.28 | −26.27 | −3.37 | Asp70, Ser151, Gly152 | 1.2953 | 100.00% |
| B5 | SLA-3*0401 | 386 | 394 | RRSRMFAPT | −32.49 | −18.96 | −0.51 | Asp70 | 0.9148 | 100.00% |
| B6 | SLA-3*0401 | 423 | 431 | RRSWQARTR | −17.93 | −20.24 | −2.17 | Arg63, Gln156, Glu167 | 0.6504 | 76.27% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Mai, J.; Yang, Y.; Wang, N. Porcine Parvovirus 7: Evolutionary Dynamics and Identification of Epitopes toward Vaccine Design. Vaccines 2020, 8, 359. https://doi.org/10.3390/vaccines8030359
Wang D, Mai J, Yang Y, Wang N. Porcine Parvovirus 7: Evolutionary Dynamics and Identification of Epitopes toward Vaccine Design. Vaccines. 2020; 8(3):359. https://doi.org/10.3390/vaccines8030359
Chicago/Turabian StyleWang, Dongliang, Jinhui Mai, Yi Yang, and Naidong Wang. 2020. "Porcine Parvovirus 7: Evolutionary Dynamics and Identification of Epitopes toward Vaccine Design" Vaccines 8, no. 3: 359. https://doi.org/10.3390/vaccines8030359
APA StyleWang, D., Mai, J., Yang, Y., & Wang, N. (2020). Porcine Parvovirus 7: Evolutionary Dynamics and Identification of Epitopes toward Vaccine Design. Vaccines, 8(3), 359. https://doi.org/10.3390/vaccines8030359