Abstract
Four decades after the eradication of smallpox, poxviruses continue to threaten the health of humans and other animals. Vaccinia virus (VACV) was used as the vaccine that successfully eradicated smallpox and is a prototypic member of the poxvirus family. Many cellular pathways play critical roles in productive poxvirus replication. These pathways provide opportunities to expand the arsenal of poxvirus antiviral development by targeting the cellular functions required for efficient poxvirus replication. In this study, we developed and optimized a secreted Gaussia luciferase-based, simplified assay procedure suitable for high throughput screening. Using this procedure, we screened a customized compound library that contained over 3200 bioactives and FDA (Food and Drug Administration)-approved chemicals, most having known cellular targets, for their inhibitory effects on VACV replication. We identified over 140 compounds that suppressed VACV replication. Many of these hits target cellular pathways previously reported to be required for efficient VACV replication, validating the effectiveness of our screening. Importantly, we also identified hits that target cellular functions with previously unknown roles in the VACV replication cycle. Among those in the latter category, we verified the antiviral role of several compounds targeting the janus kinase/signal transducer and activator of transcription-3 (JAK/STAT3) signaling pathway by showing that STAT3 inhibitors reduced VACV replication. Our findings identify pathways that are candidates for use in the prevention and treatment of poxvirus infections and additionally provide a foundation to investigate diverse cellular pathways for their roles in poxvirus replications.
1. Introduction
Smallpox, one of the deadliest diseases in human history, has claimed more human lives than all other infectious diseases combined. In the 20th century alone, more than 300 million deaths were attributed directly to smallpox [1]. Variola virus (VARV) is the causative agent of smallpox. Vaccinia virus (VACV) is a close relative of VARV, and it was the first vaccine used to eradicate smallpox [2]. However, it has been shown that the de novo assembly of infectious horsepox virus using laboratory-accessible equipment and chemically synthesized DNA is possible [3]. Concerns have been raised that a similar method could potentially be used to reconstitute VARV, which poses a public health threat because VARV from insecure stocks or assembled in a laboratory could be used as a bioweapon. Moreover, many other poxviruses continue to threaten human health in the post-smallpox era. For example, monkeypox virus, a zoonotic poxvirus genetically similar to VACV and VARV, is endemic in Central and Western Africa [4,5]. Monkeypox virus was introduced into the U.S. by exotic pet trade. The pathogen was then transmitted to humans and led to 37 confirmed cases in the U.S. in 2003 [6]. In addition, several travel-related exports of monkeypox virus have been reported in the United Kingdom, Singapore, and Israel in the past few years [7,8,9]. Molluscum contagiosum virus, another example of a human-specific poxvirus, causes a common infection throughout the world that is especially frequent in children, sexually active adults, and immunocompromised individuals [10]. Moreover, many other poxviruses, such as buffalopox and cowpox viruses in Asia and Europe, respectively, are still causing animal diseases and economic loss [11,12,13].
Though smallpox was eradicated nearly 40 years ago, the first antiviral compound to treat smallpox was only recently approved by the Food and Drug Administration (FDA) in 2018 [14,15]. The concerns for drug-resistance and adverse reactions demand the discovery of more antivirals with diverse targets [16]. Moreover, the continued surveillance and discovery of novel therapeutics for poxvirus infections are important for preventing emerging and re-emerging poxvirus pathogens other than VARV. In fact, the U.S. federal government is actively seeking additional drugs to treat smallpox and other poxvirus diseases [15,17,18]. In the past two decades, several antivirals for poxviruses have been identified, including CMX001, Tecovirimat, and CMLDBU6128 [17]. However, mutant viruses resistant to these drugs have also been identified, strongly incentivizing the identification of other new antivirals for poxvirus infections [17,18]. In addition to identifying drugs targeting viral components, an alternative approach is to discover compounds that target cellular functions required for efficient viral replication. Cellular function-targeted drugs have the potential to be broad-spectrum and are less likely to develop drug resistance [19].
In this study, we developed a Gaussia luciferase (Gluc)-based, all-in-one plate and an unbiased screening approach for compounds that can inhibit the replication of VACV. We screened a chemical library by including over 3200 compounds from Selleck bioactives and FDA-approved drugs that have known cellular targets. We identified over 140 positive hits that target various cellular processes. In addition to compounds targeting cellular functions that are unknown to be important for VACV replication such as janus kinase-signal transducer and activator of transcription-3 (JAK-STAT3) signaling, the hits also include numerous compounds targeting cellular functions that are known to be required for efficient VACV replication, thus validating the effectiveness of our screening system. Many of these positive hits reported here represent candidates for the development of cellular function-oriented poxvirus antivirals.
2. Materials and Methods
2.1. Cells and Viruses
HeLa (ATCC CCL-2) and NHDF (normal human dermal fibroblast; ATCC, Manassas, VA, USA; PCS 201-012) cells were purchased from ATCC and maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 2 mM L-glutamine, 100 units of penicillin, 100 µg of streptomycin, and 10% fetal bovine serum (FBS). BS-C-1 cells (ATCC CCL-26) were purchased from ATCC and grown in Eagle’s Minimum Essential Medium (EMEM) supplemented as described above. The recombinant VACV—Western Reserve virus expressing Gluc under viral late promoter was described elsewhere, and it was a gift from Dr. Bernard Moss [20]. Wildtype and recombinant viruses were amplified in BS-C-1 cells, and they were cushion- and gradient-purified prior to use, as described elsewhere [20].
2.2. Virus Infection
A cell culture medium from cells at a 90% confluency were removed and replaced with VACV-WR or vLGluc diluted in EMEM supplemented with 2.5% FBS. Cells were infected for one h at 37 °C. The inoculum was then discarded, and cells were washed 2× times with PBS and maintained in a fresh medium. After 24 or 48 h, viruses from each treatment were collected by freezing/thawing 3 times, or the Gluc was directly measured in the culture medium using a Glomax luminometer (Promega, Madison, WI, USA) with a Gaussia luciferase flash assay kit (ThermoFisher, Waltham, MA, USA). Virus titers were determined by plaque assay in BS-C-1 cells with viruses serially diluted in EMEM-2.5% FBS.
2.3. Gaussia Luciferase (Gluc) Reporter Screening Assay
All compounds were acoustically transferred to the assay plates using ECHO555 (Beckman Inc. Sykesville, MD, USA) at a final desired concentration. All plates included DMSO and cytosine arabinoside (AraC) controls. Cells were trypsinized, resuspended in a fresh medium, and counted before mixing with diluted reporter viruses to reach the desired multiplicity of infection (MOI). The cell–virus mixture was then plated in 96-well plates pre-coated with library drugs along with positive (AraC) and negative (DMSO) controls. The plating procedure was carried out using an automated microplate dispenser, and 50 µL of the virus–cell mixture (~5000 cells) were dispensed into each well. The plates were then incubated at 37 °C with 5% CO2. After 24 h, luciferase activities were measured using a Glomax luminometer with a Gaussia luciferase flash assay kit (Thermo Fisher 16158).
2.4. Cell Viability Assays
For high-throughput screening, HeLa cells were plated in 384-well microplates at 2000 cells/40 μL per well in DMEM containing 10% FBS. Media and vehicle control (DMSO) wells were included in each assay plate. After 24 and 48 h of incubation, cytotoxicity was measured on an Enspire plate reader (Perkin Elmer, Waltham, MA, USA) using the luminescence-based CellTiter-Glo reagent (Promega). The percent cytotoxicity was normalized to the DMSO controls.
A 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT) assay was performed using an MTT assay kit (Cayman Chemical, Ann Arbor, MI, USA, Cat#10009365) to measure the cytotoxicity of the chemical inhibitors on cells in a non-high-throughput setting. Briefly, NHDF cells in 96-well plates were treated with DMSO or chemical inhibitors dissolved in DMSO and incubated for 24 h. After incubation, 10 µL of MTT reagent were added to each well, and cells were incubated for another 3 h. Next, a 100 µL crystal resolving solution was added to each well, and absorbance at 595 nm (UV light) was measured for each well after 18 h of incubation at 37 °C.
2.5. Determination of the Effects of Compounds on Gluc Enzyme Reaction
To determine whether the compounds directly inhibited the Gaussia protein activity, we optimized a biochemical Gaussia luciferase assay counter-screen in 384-well plate format. The optimized reaction consisting of 5 nM purified Gaussia Luciferase enzyme (NanoLight Inc., Pinetop, AZ, USA) was incubated with the compounds for 10 min at room temperature before the addition of Gluc Glow Assay reagents (NanoLight Inc.). After 10 min of incubation, luminescence was measured using the Perkin Elmer Enspire, and percent activity was normalized to the negative and positive controls.
2.6. High-Throughput Screening Data Processing and Statistical Analysis
The raw luminescence data from the primary screening of 2038 bioactives and 1190 FDA-approved drug collections (Selleck Inc.) were used to calculate percent inhibition, which was normalized to the negative (DMSO) and positive (AraC) controls. The hits were identified as compounds that inhibited the luminescence to a greater extent than plate median plus three standard deviations. The average Z’ score of 0.6 ± 0.05 indicated a good separation of the positive and negative controls and the suitability of the assay for screening. The 1042 primary hits were reconfirmed in a dose response at concentrations of 20, 10, 5, and 0 mM in anti-viral assays at both low and high MOIs, and they were also evaluated at 24 and 48 h for HeLa cytotoxicity and in the counter-screen for Gaussia reporter inhibitors. A final set of 142 compounds that showed potent anti-viral activity and were not cytotoxic or Gaussia reporter inhibitors were further analyzed by cheminformatics for their modes of action.
3. Results
3.1. Development and Optimization of a Secreted Gluc-Based Assay Procedure to Screen VACV Inhibitors
We posit that Gluc is an outstanding reporter gene for a high-throughput chemical screen. It was released into the cell culture medium immediately after synthesis, which allowed it to be detected in real-time without the need to lyse the cells. Therefore, we used a reporter VACV, vLGluc, which was engineered to express Gluc in the WR strain of VACV under the control of a well-characterized viral late F17 promoter [20]. The luciferase expressed from this virus was found predominantly (99.8%) in the cell-culture medium. After extensive optimization and testing, we established a protocol for the screening, as illustrated in Figure 1A. Briefly, HeLa cells grown to >90% confluency were trypsinized, counted, and mixed with vLGluc at 0.01 PFU per cell. An automated dispenser was then used to dispense the virus–cell mixture into 96-well plates (50 µL/well), and the wells were pre-deposited with chemicals (one compound/well with desired concentration). The infection was incubated for 24 h prior to luciferase detection with a luminometer that directly injected detection reagent into the wells.
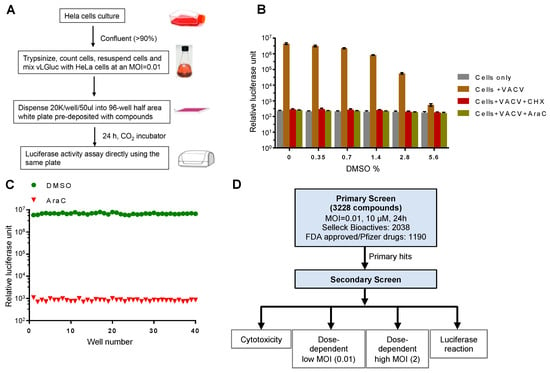
Figure 1.
Experimental design and optimization of the screening procedure. (A) Experimental procedure for the luciferase reporter assay. (B) Effect of DMSO on the reporter assay. HeLa cells were infected with vLGluc at 0.01 PFU/cell in the presence or absence of cycloheximide (CHX) or cytosine arabinoside (AraC). DMSO was added to each well to reach the indicated final concentrations (v/v). Luciferase activities were measured 24 hours post infection (hpi). Error bars indicate standard deviation (n = 3). (C) HeLa cells from 40 wells in a 96-well plate at 90% confluency were infected with vLGluc at 0.01 PFU/cell in the presence of only DMSO or AraC diluted in DMSO. Luciferase activities were determined at 24 hpi. (D) Flowchart illustrating two rounds of screenings performed. MOI: multiplicity of infection.
A screening protocol adaptable for high-throughput screening was optimized. First, in order to simplify the screening procedure as a way to reduce system variations and errors, we made changes to the typical VACV infection protocol of adherent cells by mixing cell suspension and viruses prior to plating cells in multi-well plates containing controls and compounds. Second, the Gaussia luciferase detection reagent was directly injected into the wells with infected cells at 24 h post infection (hpi) to eliminate variability resulting from media transfer to new plates. Third, we used a low MOI, which ensured the discovery of chemicals working on all steps of VACV replication and spread. Finally, the highly sensitive glow type Gluc signal was quite stable for detection. All steps were amenable to automated liquid dispensing. In this procedure, to counteract discrepancies caused by plate-to-plate variation, we included negative control wells containing DMSO only, which was used to dissolve drugs. We also included positive control wells with AraC, a well-established chemical to block VACV DNA replication and downstream post-replicative gene expression [21]. To test the effect of DMSO on Gluc expression from the virus, we performed a dose-dependent experiment using various DMSO concentrations. We found a minimal influence of DMSO at the concentration we used (<0.35%) for the screening (Figure 1B). Moreover, we optimized cell density (90% confluency), medium volume (50 µL/well), MOI (0.01), and incubation time (24 h) to increase the reproducibility of the assay. As a result, we were able to establish a very low well-to-well variation using AraC in the pilot test (Figure 1C).
Next, we assembled a customized drug library that contains 3228 compounds. This library includes drugs from the Selleck Bioactives and FDA-approved chemicals. One advantage of this library is that most of these compounds have at least one known cellular target, which would help identify new cellular processes/pathways utilized by VACV to facilitate its own replication. The library also provides redundancy because multiple compounds often target a common cellular target. The library was screened for the drugs’ inhibition on the expression of the Gluc reporter gene in a procedure illustrated in Figure 1D. For the primary screen, we screened the drugs with a low MOI to monitor their effect on spread using the procedure shown in Figure 1A. The positive hits were selected based on their ability to reduce luciferase production as compared to AraC (100%). For the secondary screen, we tested the primary hits using different doses of chemicals (5, 10, and 20 µM) at both low and higher MOIs (0.01 and 2, respectively). We also tested the cytotoxicity of the chemicals on HeLa cells and their direct effects on purified Gluc enzyme activity/reaction to exclude false positives (Figure 1D).
3.2. Identification of VACV Inhibitors Targeting a Broad Range of Cellular Targets and Processes
We normalized all hits to the negative and positive controls where DMSO treatment alone was set to 0% and AraC treatment was set to 100% luciferase inhibition. All hits were plotted by their effects on Gluc production in the primary screen at 10 µM in Figure 2A. We found that most of the hits showed the inhibition of VACV replication at MOIs of 0.01 and 2. Some of the inhibitors had more potent effects of reducing VACV replication at the MOI of 0.01 (Figure 2B), thus suggesting that cell-to-cell spread was also affected by these hits. The majority of the hits showed a low cytotoxicity compared to DMSO, the solvent used for all drugs (Figure 2C). In addition, the positive effects we observed were not a result of the direct inhibition of the enzymatic activity or reaction of the Gluc (Figure 2D). To further characterize the inhibitors, we performed a dose-dependent study in which we included two more dosages (5 and 20 µM) in addition to the initial dose (10 µM). We found that most of them showed a dose-dependent inhibition of VACV replication, indicating a specific effect of the chemicals on VACV replication (Figure 2E). Overall, after the procedure, we identified 142 hits that significantly reduced Gluc activities in the viral reporter assay.
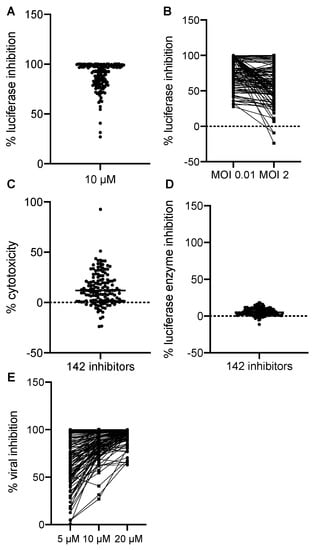
Figure 2.
Screening results. (A) The effects of 142 positive hits at 10 µM were plotted. Each dot represents one compound. Luciferase activity from each well was normalized to that of the positive control wells containing AraC (100%) diluted in DMSO. (B) The inhibitory effect of all 142 positive hits at low (0.01) and high (2) MOIs were plotted. The % luciferase inhibition was calculated as described in (A). (C) Cytotoxic effects of all 142 positive hits were examined. DMSO’s effect was set to 0%, and the data of all hits were normalized to DMSO only. (D) The effect of positive hits on the luciferase reaction itself was examined by incubating the compounds with a purified Gaussia luciferase enzyme, and luciferase activity was measured after 10 min of incubation. Data from DMSO-only wells were set to 0%. (E) Dose-dependent effects of 142 hits were plotted. Data were acquired and normalized as described above.
All 142 positive hits and their cellular targets are listed and clustered by their common cellular functions in Table 1. The most apparent category was nucleic acid analogs, which were predicted to have antiviral functions because they could be integrated into actively replicating DNA and could terminate DNA synthesis. The nucleic acid analogs would usually block cellular DNA replication in a similar mechanism; however, at the tested concentrations, the analogs showed no or low toxic effects on cell viability. The most abundant positive hits were found in the categories of HSP90 (heat shock protein 90) and EGFR (epidermal growth factor receptor) inhibitors, both of which have been previously shown to be required for efficient VACV replication [22,23]. In addition, microtubules, mTOR (mammalian target of rapamycin), proteasome, and CRM1 (chromosomal maintenance 1) have also been shown to be indispensable for VACV replication in cells by other techniques [24,25,26,27,28,29,30]. The fact that we were able to pick up many positive hits in these pathways known to be crucial for VACV replication demonstrated our screening method’s high effectiveness on identifying bona fide VACV inhibitors.

Table 1.
Vaccinia virus (VACV) inhibitors identified after two-round of screens. FDA: Food and Drug Administration.
Remarkably, we also identified many hits that target biological processes that were previously unknown to promote VACV replication. Examples of those include inhibitors of the JAK-STAT pathway, topoisomerase II, and SIRT1 (sirtuin 1). The identification of these chemicals opens exciting new directions for understanding their roles in facilitating VACV replication and additional cellular targets for the development of new antivirals.
Interestingly, 35 of the identified positive hits were drugs already approved by the FDA for antiviral or non-antiviral-purposes. For example, floxuridine was approved in the 1970s for cancer treatment, and it belongs to the class of antimetabolites [67]. In addition, anagrelide is a drug used to treat essential thrombocytosis and chronic myeloid leukemia. It works by inhibiting the maturation of platelets from megakaryocytes [68]. Moreover, desmethyl erlotinib is an EGFR inhibitor, and it was approved in 2004 to treat lung and pancreatic cancer [69]. All FDA-approved drugs identified have some known modes of actions and have shown the potent inhibition of VACV in the screens (Table 1), suggesting a promising direction of exploring novel and safe antivirals for poxvirus infection.
3.3. Verification of the Suppressing Effects of JAK-STAT3 Pathway Inhibitors on VACV Replication
Among the positive hits, we noticed multiple compounds with a known function to inhibit the JAK-STAT pathway. The JAK-STAT signaling pathway is known to participate in various cellular processes such as immunity, cell division, cell death, and tumorigenesis [70]. The pathway is comprised of three major parts: receptors that could react to stimulations from different ligands under different physiological conditions, JAKs, and STAT proteins [70]. The disruption of the JAK-STAT pathway leads to a variety of diseases including cancer, immune disorders, and skin conditions [71]. In humans, there are four JAKs (JAK1, JAK2, JAK3, and TYK2) and seven STATs (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6). The combination of different JAKs and STATs could result in the activation and/or inactivation of different cellular processes through transcriptional regulation [71,72]. The specific targets of the identified positive hits are listed in Table 2. Interestingly, among all JAK-STAT inhibitors, STAT3 was a convergent target found in our screening.
Table 2.
JAK-STAT3 inhibitors identified after two-round of screens.
To verify the effects of inhibitors on the JAK-STAT3 pathway in the VACV life cycle, we decided to first repeat the reporter assay with three compounds identified from the screen including SC144, AZ960, and niclosamide, each of which targets one component of the pathway, namely the ligand-receptor (SC144), the JAK2 kinase (AZ960), and the STAT3 protein (niclosamide). Additionally, we included a widely reported and selective STAT3 inhibitor static that was not picked up in our screening. We also chose to test the inhibitors in a different cell type: NHDF, which is a line of human dermal fibroblasts of non-cancerous origin. After optimizing the working concentrations for the drugs in NHDF cells (niclosamide and SC144: 1 µM, stattic: 3 µM), we determined late gene expression by infecting the cells with the same reporter virus (vLGluc) used in the drug treatment screenings, and we monitored luciferase activities at 8 hpi. Our analysis showed that all drugs significantly suppressed viral late gene expression without negatively affecting cell viability at the indicated concentrations (Figure 3A,B). Next, we sought to examine if blocking STAT3 with chemical inhibitors could directly impair viral replication. NHDF cells were treated with DMSO, niclosamide, AZ960, SC144, or stattic for one hour before they were infected with VACV-WR at 0.01 PFU/cell. Viruses were harvested at 48 hpi, and virus titers were determined by a plaque assay in BS-C-1 cells. As shown in Figure 3C, all four tested STAT3 inhibitors substantially repressed VACV replication, indicating that blocking STAT3 diminished VACV replication in human cells.
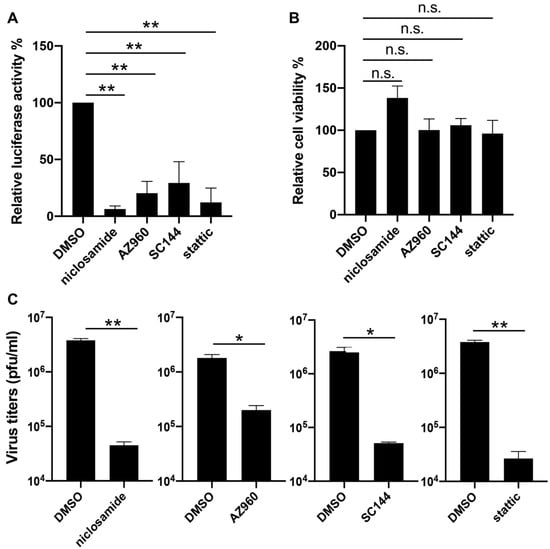
Figure 3.
Inhibition of VACV by STAT3 inhibitors. (A) Normal human dermal fibroblast (NHDF) cells were treated with DMSO, niclosamide (1 µM), AZ960 (3 µM), SC144 (1 µM), or static (3 µM), and then they were infected with vLGluc at 1 PFU/cell. At 8 h later, luciferase activity was determined in collected cell culture media. (B) NHDF cells were treated with the above drugs for 24 h and cell viability was measured by an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide) assay. Three biological replicates were carried out. Error bars indicated standard deviation. (C) NHDF cells were treated with selected STAT3 inhibitors at the concentrations mentioned above and infected with VACV-WR at 0.01 PFU/cell. Viruses were collected at 48 hpi and tittered in BS-C-1 cells. Error bars indicate standard deviation (n = 3). Statistics: * p < 0.05, ** p < 0.01, n.s.: non-significant by two-sided Students’ T test.
4. Discussion
After a series of optimizations, including adjustments to parameters such as cell number, infection time, and infection MOI, we developed an assay to screen anti-VACV compounds with all steps performed in one single plate. We screened a library with over 3200 compounds by performing multiple rounds of screening for their effects on virus spread, cell toxicity, and on the reporter assay itself at various doses, and we identified more than 140 positive hits that showed the statistically significant inhibition of VACV spread but manageable levels of toxicity on cell viability. The assay developed here could be readily scaled up for screening with a much higher capacity.
The utility and effectiveness of this assay were validated by the discovery of positive hits that targeted cellular pathways that were reported to be important for VACV replication. For example, Hung et al. reported that HSP90 redistributed upon VACV infection and a chemical geldanamycin (GA), which blocks the ATPase activity of HSP90, strongly suppressed virus infection in RK13 cells [22]. Moreover, Kowalczyk and coworkers observed that both HSP70 and HSP90 translocated to the nucleus at the early time of VACV infection, and the mRNA abundance of HSP90 was perturbed by the virus infection [73]. These results suggest that HSP90 may promote VACV replication. We found 12 different compounds in our screen that were previously known to inhibit HSP90, which further emphasized the importance of HSP90 in supporting VACV infection and encouraged more studies at the molecular level. It is possible that HSP90 assists the folding of VACV virion proteins for proper virus assembly. However, the actual mechanism needs more thorough investigations. Another example is the mTOR-related pathways. The mTOR is a key regulator of many cellular pathways that can respond to both extracellular and intracellular stimuli [74]. Many viruses have evolved strategies to promote, reduce, or even inhibit the mTOR pathway for their own replication [75]. It has been shown that VACV activates the mTOR pathway to delay apoptotic signals, which promote viral replication in mammalian cells [25]. However, it was also shown that the F17 protein made late during a VACV infection was able to dysregulate the activity of mTOR, which paralyzed its role in activating STING (stimulator of interferon genes) and the downstream antiviral responses [25]. In our screen, we found seven inhibitors of the PI3K/mTOR pathway that showed the significant suppression of VACV replication. Again, these findings demonstrated the effectiveness of our screening assay.
Importantly, we identified many compounds that target cellular functions that have not been previously reported as necessary for VACV replication including aryl hydrocarbon receptor (AhR), cytochrome P450, and aurora kinases A/B. Many of these pathways have been extensively studied in other fields, and tools are readily available for the manipulation for some of them. However, their involvement in poxvirus infections has not been previously reported. Admittedly, viruses are obligate intracellular parasites, and their reproduction heavily depends on the host cell’s machinery and metabolism. It is expected that the disruption of many essential cellular functions by chemicals or genetic alterations could lead to the arrest of cellular functions that will inevitably jeopardize virus replication. However, it is worth noting that all the positive hits reported here were tested for their effect on cell viability for up to 48 h at various concentrations and at concentrations where they exhibit the vigorous inhibition of virus gene expression, and most of them showed no or very low negative effects on cell viability. A possible explanation is that the virus might be capable of temporarily ramping up some cellular pathways to degrees that are only beneficial to the virus but superfluous to the cells. The chemical inhibition of those pathways would pose a more significant effect on viral replication than on cell metabolism. These pathways are, therefore, outstanding candidates for targeted antiviral therapy using chemical or genetic approaches. Our screening has provided multiple candidates for future research and validation, especially the thirty-five FDA-approved drugs.
We performed further analyses with inhibitors of the JAK-STAT3 pathway in primary cells. Our results confirmed that the inhibition of components diminished virus replication and late gene expression, suggesting a role of STAT3 in promoting VACV replication in human cells. Interestingly, another study published several years ago investigated the function of STAT3 in VACV-ACAM2000 infection, and the results suggested a defensive role of STAT3 in VACV infection [76]. It is worth noting that the studies differed in two key aspects, which may explain the observed differences. First, while the previous study was performed in human and mouse keratinocytes, we executed our screens in HeLa cells and confirmed our findings in NHDFs. Secondly, the prior study used the ACAM2000 strain of VACV, a clone derived from Dryvax [77]. ACAM2000 is a live attenuated strain approved as a smallpox vaccine [78]. VACV-WR, the virus used in our study, is, on the other hand, one of the most virulent VACV strains tested in animal models and was considered unsuitable to be used as a vaccine due to its high neuropathogenicity in mice models [79]. A comparison between the genomes of VACV-WR and ACAM2000 showed the ACAM2000 has two major deletions in the inverted terminal repeats (ITR, C12-C18 at the left ITR and the corresponding duplicates at the right ITR), as well as mutations throughout the genome [77,80]. Among those deleted, C12 has been shown to be important for enabling modified vaccinia virus Ankara (MVA) replication in human cells [81,82]. Some of those genes lost in ACAM2000 might regulate innate immune responses in cells, which complicates the role of STAT3 in VACV replication. Further investigations are needed to define the role of STAT3 in the replication of different VACV strains and elucidate the mechanism involved in the requirement of STAT3 signaling VACV replication.
Antivirals are indispensable therapeutics in preparing for emerging and re-emerging infectious diseases. They could be used post-virus exposure in patients and could work rapidly in constraining virus dissemination compared to vaccines. Our study has expanded the pool of therapeutic candidates for future pre-clinical and clinical tests to treat poxvirus infections. Further preclinical screening of the candidates listed in this study will be needed to evaluate drug effects in animal models, pharmacokinetics, the development of drug resistance, and in vivo toxicity before the most likely effective antiviral compounds for further clinical evaluation can be identified. Additionally and importantly, we identified new cell targets for understanding their roles in poxvirus infections and as targets for new antiviral development, which is complementary to a few previous genetic screens of cellular genes needed for efficient VACV replication [29,30,83].
5. Conclusions
Using a Gaussia luciferase-based reporter assay suitable for high-throughput screening, we identified over 140 compounds that could suppress VACV replication and spread in cultured human cells. These compounds are from a chemical library containing bioactives and FDA-approved drugs with known cellular targets. These cellular functions targeted by the positive hits include those previously known to be important for VACV replication, as well as those with previously unknown roles in supporting VACV replication. Further, we verified the inhibitory effects of multiple JAK-STAT3 inhibitors on viral replication. Our results provide promising candidate compounds for future evaluation in treating orthopoxvirus infections. The cell functions targeted by these compounds need additional investigations for understanding their roles in poxvirus infections.
Author Contributions
Conceptualization, Z.Y.; methodology, C.P., A.R., and Z.Y.; formal analysis, C.P., A.R., and Z.Y.; investigation, C.P., Y.Z., S.C., A.P., M.L.C.G., P.M., and A.R; data curation, C.P., A.R., and Z.Y.; writing—original draft preparation, C.P.; writing—review and editing, C.P., A.R., and Z.Y.; visualization, C.P. and A.R.; supervision, Z.Y.; funding acquisition, Z.Y. and A.R. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this publication was supported, in part, by the National Institute of General Medical Sciences (NIGMS, award number P20GM113117), and National Institute of Allergy and Infectious Diseases (NIAID, award number R01AI143709), of the National Institutes of Health under award number. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Bernard Moss from the NIH for providing reagents. We thank Mark Gray for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Chapter 1: Smallpox: Eradicating an ancient scourge. In Bugs, Drugs and Smoke; WHO Press: Geneva, Switzerland, 2011; pp. 3–21. [Google Scholar]
- Moss, B. Poxviridae, Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2129–2159. [Google Scholar]
- Noyce, R.S.; Lederman, S.; Evans, D.H. Construction of an infectious horsepox virus vaccine from chemically synthesized DNA fragments. PLoS ONE 2018, 13, e0188453. [Google Scholar] [CrossRef] [PubMed]
- Beer, E.M.; Rao, V.B. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLoS Negl. Trop. Dis. 2019, 13, e0007791. [Google Scholar] [CrossRef] [PubMed]
- Petersen, E.; Kantele, A.; Koopmans, M.; Asogun, D.; Yinka-Ogunleye, A.; Ihekweazu, C.; Zumla, A. Human Monkeypox: Epidemiologic and Clinical Characteristics, Diagnosis, and Prevention. Infect. Dis. Clin. N. Am. 2019, 33, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Update: Multistate outbreak of monkeypox—Illinois, Indiana, Kansas, Missouri, Ohio, and Wisconsin, 2003. MMWR Morb. Mortal Wkly Rep. 2003, 52, 642–646. [Google Scholar]
- Vaughan, A.; Aarons, E.; Astbury, J.; Balasegaram, S.; Beadsworth, M.; Beck, C.R.; Chand, M.; O’Connor, C.; Dunning, J.; Ghebrehewet, S.; et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveillance 2018, 23, 1800509. [Google Scholar] [CrossRef] [PubMed]
- Ng, O.T.; Lee, V.; Marimuthu, K.; Vasoo, S.; Chan, G.; Lin, R.T.P.; Leo, S.Y. A case of imported Monkeypox in Singapore. Lancet Infect. Dis. 2019, 19, 1166. [Google Scholar] [CrossRef]
- Erez, N.; Achdout, H.; Milrot, E.; Schwartz, Y.; Wiener-Well, Y.; Paran, N.; Politi, B.; Tamir, H.; Israely, T.; Weiss, S.; et al. Diagnosis of Imported Monkeypox, Israel, 2018. Emerg. Infect. Dis. 2019, 25, 980–983. [Google Scholar] [CrossRef]
- Meza-Romero, R.; Navarrete-Dechent, C.; Downey, C. Molluscum contagiosum: An update and review of new perspectives in etiology, diagnosis, and treatment. Clin. Cosmet. Investig. Dermatol. 2019, 12, 373–381. [Google Scholar] [CrossRef]
- Lewis-Jones, S. Zoonotic poxvirus infections in humans. Curr. Opin. Infect. Dis. 2004, 17, 81–89. [Google Scholar] [CrossRef]
- Oliveira, G.P.; Rodrigues, R.A.L.; Lima, M.T.; Drumond, B.P.; Abrahão, J.S. Poxvirus Host Range Genes and Virus-Host Spectrum: A Critical Review. Viruses 2017, 9, 331. [Google Scholar] [CrossRef]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Merchlinsky, M.; Albright, A.; Olson, V.; Schiltz, H.; Merkeley, T.; Hughes, C.; Petersen, B.; Challberg, M. The development and approval of tecoviromat (TPOXX®), the first antiviral against smallpox. Antivir. Res. 2019, 168, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Chan-Tack, K.M.; Harrington, P.R.; Choi, S.Y.; Myers, L.; O’Rear, J.; Seo, S.; McMillan, D.; Ghantous, H.; Birnkrant, D.; Sherwat, A.I. Assessing a drug for an eradicated human disease: US Food and Drug Administration review of tecovirimat for the treatment of smallpox. Lancet Infect. Dis. 2019, 19, e221–e224. [Google Scholar] [CrossRef]
- Pires, M.A.; Rodrigues, N.F.S.; de Oliveira, D.B.; de Assis, F.L.; Costa, G.B.; Kroon, E.G.; Mota, B.E.F. In vitro susceptibility to ST-246 and Cidofovir corroborates the phylogenetic separation of Brazilian Vaccinia virus into two clades. Antivir. Res. 2018, 152, 36–44. [Google Scholar] [CrossRef]
- Delaune, D.; Iseni, F. Drug Development against Smallpox: Present and Future. Antimicrob. Agents Chemother. 2020, 64, e01683–e01719. [Google Scholar] [CrossRef]
- Duraffour, S.; Andrei, G.; Topalis, D.; Krečmerová, M.; Crance, J.M.; Garin, D.; Snoeck, R. Mutations conferring resistance to viral DNA polymerase inhibitors in camelpox virus give different drug-susceptibility profiles in vaccinia virus. J. Virol. 2012, 86, 7310–7325. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Pant, A.; Cao, S.; Yang, Z. Asparagine Is a Critical Limiting Metabolite for Vaccinia Virus Protein Synthesis during Glutamine Deprivation. J. Virol. 2019, 93, e01834–e01918. [Google Scholar] [CrossRef]
- Renis, H.E.; Johnson, H.G. Inhibition of plaque formation of vaccinia virus by cytosine arabinoside hydrochloride. Bacteriol. Proc. 1962, 45, 45. [Google Scholar]
- Hung, J.J.; Chung, C.S.; Chang, W. Molecular chaperone Hsp90 is important for vaccinia virus growth in cells. J. Virol. 2002, 76, 1379–1390. [Google Scholar] [CrossRef]
- Beerli, C.; Yakimovich, A.; Kilcher, S.; Reynoso, G.V.; Fläschner, G.; Müller, D.J.; Hickman, H.D.; Mercer, J. Vaccinia virus hijacks EGFR signalling to enhance virus spread through rapid and directed infected cell motility. Nat. Microbiol. 2019, 4, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Way, M. The role of signalling and the cytoskeleton during Vaccinia Virus egress. Virus Res. 2015, 209, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Meade, N.; King, M.; Munger, J.; Walsh, D. mTOR Dysregulation by Vaccinia Virus F17 Controls Multiple Processes with Varying Roles in Infection. J. Virol. 2019, 93, e00784–e00819. [Google Scholar] [CrossRef] [PubMed]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143–1157. [Google Scholar] [CrossRef]
- Slezak, K.; Michalik, M.; Kowalczyk, A.; Rokita, H. YY1 is recruited to the cytoplasm of vaccinia virus-infected human macrophages by the Crm1 system. Virus Res. 2004, 102, 177–784. [Google Scholar] [CrossRef]
- Satheshkumar, P.S.; Anton, L.C.; Sanz, P.; Moss, B. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 2009, 83, 2469–2479. [Google Scholar] [CrossRef]
- Sivan, G.; Martin, S.E.; Myers, T.G.; Buehler, E.; Szymczyk, K.H.; Ormanoglu, P.; Moss, B. Human genome-wide RNAi screen reveals a role for nuclear pore proteins in poxvirus morphogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3519–3524. [Google Scholar] [CrossRef]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.E.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi screening reveals proteasome- and Cullin3-dependent stages in vaccinia virus infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Maluquer de Motes, C.; Smith, G.L. Vaccinia virus protein A49 activates Wnt signalling by targetting the E3 ligase β-TrCP. J. Gen. Virol. 2017, 98, 3086–3092. [Google Scholar] [CrossRef]
- Santos, C.R.; Vega, F.M.; Blanco, S.; Barcia, R.; Lazo, P.A. The vaccinia virus B1R kinase induces p53 downregulation by an Mdm2-dependent mechanism. Virology 2004, 328, 254–265. [Google Scholar] [CrossRef]
- Remichkova, M.; Petrov, N.; Galabov, A.S. Synergistic Combination Effect of Cidofovir and Idoxuridine on Vaccinia Virus Replication. Antivir. Chem. Chemother. 2016, 17, 53–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moss, B. Poxvirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010199. [Google Scholar] [CrossRef] [PubMed]
- Smee, D.F. Orthopoxvirus inhibitors that are active in animal models: An update from 2008 to 2012. Future Virol. 2013, 8, 891–901. [Google Scholar] [CrossRef]
- Gammon, D.B.; Gowrishankar, B.; Duraffour, S.; Andrei, G.; Upton, C.; Evans, D.H. Vaccinia virus-encoded ribonucleotide reductase subunits are differentially required for replication and pathogenesis. PLoS Pathog. 2010, 6, e1000984. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Franke, C.A.; Hruby, D.E. Inhibition of vaccinia virus replication by nicotinamide: Evidence for ADP-ribosylation of viral proteins. Virus Res. 1988, 9, 119–132. [Google Scholar] [CrossRef]
- Ryerson, M.R.; Richards, M.M.; Kvansakul, M.; Hawkins, C.J.; Shisler, J.L. Vaccinia Virus Encodes a Novel Inhibitor of Apoptosis That Associates with the Apoptosome. J. Virol. 2017, 91, e01385–e01417. [Google Scholar] [CrossRef] [PubMed]
- Baixeras, E.; Cebrián, A.; Albar, J.P.; Salas, J.; Martínez, A.C.; Viñuela, E.; Revilla, Y. Vaccinia virus-induced apoptosis in immature B lymphocytes: Role of cellular Bcl-2. Virus Res. 1998, 58, 107–113. [Google Scholar] [CrossRef]
- Piróg, K.A.; Kowalczyk, A.K.; Rokita, H.B. Changes in Bcl-2 expression in vaccinia virus-infected human peripheral blood monocytes. Viral Immunol. 2005, 18, 224–231. [Google Scholar] [CrossRef]
- Liu, Y.; Wolff, K.C.; Jacobs, B.L.; Samuel, C.E. Vaccinia virus E3L interferon resistance protein inhibits the interferon-induced adenosine deaminase A-to-I editing activity. Virology 2001, 289, 378–387. [Google Scholar] [CrossRef]
- Connor, J.D.; Sweetman, L.; Carey, S.; Stuckey, M.A.; Buchanan, R. Effect of adenosine deaminase upon the antiviral activity in vitro of adenine arabinoside for vaccinia virus. Antimicrob. Agents Chemother. 1974, 6, 630–636. [Google Scholar] [CrossRef][Green Version]
- Chen, R.A.J.; Ryzhakov, G.; Cooray, S.; Randow, F.; Smith, G.L. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008, 4, e22. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Stuart, J.H.; Talbot-Cooper, C.; Agrawal-Singh, S.; Huntly, B.; Smid, A.I.; Snowden, J.S.; Dupont, L.; Smith, G.L. Histone deacetylase 4 promotes type I interferon signaling, restricts DNA viruses, and is degraded via vaccinia virus protein C6. Proc. Natl. Acad. Sci. USA 2019, 116, 11997–12006. [Google Scholar] [CrossRef] [PubMed]
- Greseth, M.D.; Traktman, P. De novo fatty acid biosynthesis contributes significantly to establishment of a bioenergetically favorable environment for vaccinia virus infection. PLoS Pathog. 2014, 10, e1004021. [Google Scholar] [CrossRef] [PubMed]
- Reading, P.C.; Moore, J.B.; Smith, G.L. Steroid hormone synthesis by vaccinia virus suppresses the inflammatory response to infection. J. Exp. Med. 2003, 197, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Carter, G.C.; Rodger, G.; Murphy, B.J.; Law, M.; Krauss, O.; Hollinshead, M.; Smith, G.L. Vaccinia virus cores are transported on microtubules. J. Gen. Virol. 2003, 84, 2443–2458. [Google Scholar] [CrossRef]
- Ward, B.M.; Moss, B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 2001, 75, 11651–11663. [Google Scholar] [CrossRef]
- Rietdorf, J.; Ploubidou, A.; Reckmann, I.; Holmström, A.; Frischknecht, F.; Zettl, M.; Zimmermann, T.; Way, M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 2001, 3, 992–1000. [Google Scholar] [CrossRef]
- Hollinshead, M.; Rodger, G.; Van Eijl, H.; Law, M.; Hollinshead, R.; Vaux, D.J.; Smith, G.L. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 2001, 154, 389–402. [Google Scholar] [CrossRef]
- Bonjardim, C.A. Viral exploitation of the MEK/ERK pathway—A tale of vaccinia virus and other viruses. Virology 2017, 507, 267–275. [Google Scholar] [CrossRef]
- Torres, A.A.; Albarnaz, J.D.; Bonjardim, C.A.; Smith, G.L. Multiple Bcl-2 family immunomodulators from vaccinia virus regulate MAPK/AP-1 activation. J. Gen. Virol. 2016, 97, 2346–2351. [Google Scholar] [CrossRef]
- Andrade, A.A.; Silva, P.N.G.; Pereira, A.C.T.C.; De Sousa, L.P.; Ferreira, P.C.P.; Gazzinelli, R.T.; Kroon, E.G.; Ropert, C.; Bonjardim, C.A. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 2004, 381, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Rizopoulos, Z.; Balistreri, G.; Kilcher, S.; Martin, C.K.; Syedbasha, M.; Helenius, A.; Mercer, J. Vaccinia Virus Infection Requires Maturation of Macropinosomes. Traffic 2015, 16, 814–831. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, R.; Rogers, E.; Murooka, T.; Kislinger, T.; Fish, E.N. Glomulin: A permissivity factor for vaccinia virus infection. J. Interferon Cytokine Res. 2012, 32, 127–137. [Google Scholar] [CrossRef]
- Singh, P.; Yao, Y.; Weliver, A.; Broxmeyer, H.E.; Hong, S.C.; Chang, C.H. Vaccinia virus infection modulates the hematopoietic cell compartments in the bone marrow. Stem Cells 2008, 26, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.G.G.; Torres, A.A.; De Oliveira, L.C.; Da Cruz, A.F.P.; Soares-Martins, J.A.P.; Pereira, A.C.T.C.; Trindade, G.S.; Abrahao, J.S.; Kroon, E.G.; Ferreira, P.C.P.; et al. c-Jun integrates signals from both MEK/ERK and MKK/JNK pathways upon vaccinia virus infection. Arch. Virol. 2017, 162, 2971–2981. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Hofstetter, W.; Guo, W.; Li, H.; Pataer, A.; Peng, H.H.; Huo, Z.S.; Bartlett, D.L.; Lin, A.; Swisher, S.G.; et al. JNK-deficiency enhanced oncolytic vaccinia virus replication and blocked activation of double-stranded RNA-dependent protein kinase. Cancer Gene Ther. 2008, 15, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Izmailyan, R.; Hsao, J.C.; Chung, C.S.; Chen, C.H.; Hsu, P.W.C.; Liao, C.L.; Chang, W. Integrin β1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 2012, 86, 6677–6687. [Google Scholar] [CrossRef] [PubMed]
- McNulty, S.; Bornmann, W.; Schriewer, J.; Werner, C.; Smith, S.K.; Olson, V.A.; Damon, I.K.; Buller, R.M.; Heuser, J.; Kalman, D. Multiple phosphatidylinositol 3-kinases regulate vaccinia virus morphogenesis. PLoS ONE 2010, 5, e10884. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.A.P.; Leite, F.G.G.; Andrade, L.G.; Torres, A.A.; De Sousa, L.P.; Barcelos, L.S.; Teixeira, M.M.; Ferreira, P.C.P.; Kroon, E.G.; Souto-Padron, T.; et al. Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J. Virol. 2009, 83, 6883–6899. [Google Scholar] [CrossRef]
- Zaborowska, I.; Walsh, D. PI3K signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J. Virol. 2009, 83, 3988–3992. [Google Scholar] [CrossRef]
- Newsome, T.P.; Weisswange, I.; Frischknecht, F.; Way, M. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell Microbiol. 2006, 8, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Newsome, T.P.; Scaplehorn, N.; Way, M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 2004, 306, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.I.; Bleck, C.K.E.; Reh, L.; Novy, K.; Wollscheid, B.; Helenius, A.; Stahlberg, H.; Mercer, J. Vaccinia virus entry is followed by core activation and proteasome-mediated release of the immunomodulatory effector VH1 from lateral bodies. Cell Rep. 2013, 4, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.J.; Li, J.; Irwin, C.R.; Jenkins, H.; DeLange, L.; Evans, D.H. Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J. Virol. 2008, 82, 5922–5932. [Google Scholar] [CrossRef]
- Allen-Mersh, T.G.; Earlam, S.; Fordy, C.; Abrams, K.; Houghton, J. Quality of life and survival with continuous hepatic-artery floxuridine infusion for colorectal liver metastases. Lancet 1994, 344, 1255–1260. [Google Scholar] [CrossRef]
- Mazzucconi, M.G.; Baldacci, E.; Latagliata, R.; Breccia, M.; Paoloni, F.; Di Veroli, A.; Cedrone, M.; Anaclerico, B.; Villivà, N.; Porrini, R.; et al. Anagrelide in Essential Thrombocythemia (ET): Results from 150 patients over 25 years by the ’Ph1-negative Myeloproliferative Neoplasms Latium Group’. Eur. J. Haematol. 2020, ejh.13454. [Google Scholar] [CrossRef]
- Kulkarni, N.S.; Parvathaneni, V.; Shukla, S.K.; Barasa, L.; Perron, J.C.; Yoganathan, S.; Muth, A.; Gupta, V. Tyrosine kinase inhibitor conjugated quantum dots for non-small cell lung cancer (NSCLC) treatment. Eur. J. Pharm. Sci. 2019, 133, 145–159. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 3, 900–911. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- Kowalczyk, A.; Guzik, K.; Slezak, K.; Dziedzic, J.; Rokita, H. Heat shock protein and heat shock factor 1 expression and localization in vaccinia virus infected human monocyte derived macrophages. J. Inflamm. (Lond.) 2005, 2, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, V.; Cinti, A.; Amorim, R.; Mouland, A.J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 2016, 8, 152. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fisher, R.; Chowdhury, S.; Sultana, I.; Pereira, C.P.; Bray, M. Vaccinia virus induces rapid necrosis in keratinocytes by a STAT3-dependent mechanism. PLoS ONE 2014, 9, e113690. [Google Scholar] [CrossRef]
- Osborne, J.D.; Da Silva, M.; Frace, A.M.; Sammons, S.A.; Olsen-Rasmussen, M.; Upton, C.; Buller, R.M.L.; Chen, N.; Feng, Z.; Roper, R.L.; et al. Genomic differences of Vaccinia virus clones from Dryvax smallpox vaccine: The Dryvax-like ACAM2000 and the mouse neurovirulent Clone-3. Vaccine 2007, 25, 8807–8832. [Google Scholar] [CrossRef]
- Melamed, S.; Wyatt, L.S.; Kastenmayer, R.J.; Moss, B. Attenuation and immunogenicity of host-range extended modified vaccinia virus Ankara recombinants. Vaccine 2013, 31, 4569–4577. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef]
- Prazsák, I.; Tombácz, D.; Szűcs, A.; Dénes, B.; Snyder, M.; Boldogkői, Z. Full Genome Sequence of the Western Reserve Strain of Vaccinia Virus Determined by Third-Generation Sequencing. Genome Announc. 2018, 6, e01570–e01617. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Mendez-Rios, J.D.; Peng, C.; Xiao, W.; Weisberg, A.S.; Wyatt, L.S.; Moss, B. SPI-1 is a missing host-range factor required for replication of the attenuated modified vaccinia Ankara (MVA) vaccine vector in human cells. PLoS Pathog. 2019, 15, e1007710. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Moss, B. Repair of a previously uncharacterized second host-range gene contributes to full replication of modified vaccinia virus Ankara (MVA) in human cells. Proc. Natl. Acad. Sci. USA 2020, 117, 3759–3767. [Google Scholar] [CrossRef] [PubMed]
- Moser, T.S.; Jones, R.G.; Thompson, C.B.; Coyne, C.B.; Cherry, S. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathog. 2010, 6, e1000954. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).