Proteomic Analysis of Human Immune Responses to Live-Attenuated Tularemia Vaccine


 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Protein Spike-in Controls
2.3. Shared Protein Sequence Database and Protein Families
2.4. LC-MS/MS Proteomics Experiment and Data Processing
2.5. 2D-DIGE/MS Proteomics Experiment and Data Processing
2.6. Statistical Analysis
2.6.1. Normalization
- (1)
- for each sample, the median of the log2 protein signal distribution was determined.
- (2)
- the global median of all 30 sample medians calculated in (1) was obtained.
- (3)
- a sample specific scaling factor was then calculated as the difference (log2 scale) between the global median obtained in (2) and the sample-specific median obtained in (1).
- (4)
- the log2 protein signal distribution for each sample was then median normalized by adding the scaling factor (log2 scale), determined in (4).
2.6.2. Missing Value Imputation and Log Fold Change from Baseline Calculation
2.6.3. Identification of Differentially Abundant Proteins
2.6.4. Pathway Enrichment Analysis
2.6.5. Identification of Protein Responses that Best Predicted Humoral and Cellular Responses
2.7. Transcriptomics Data
3. Results
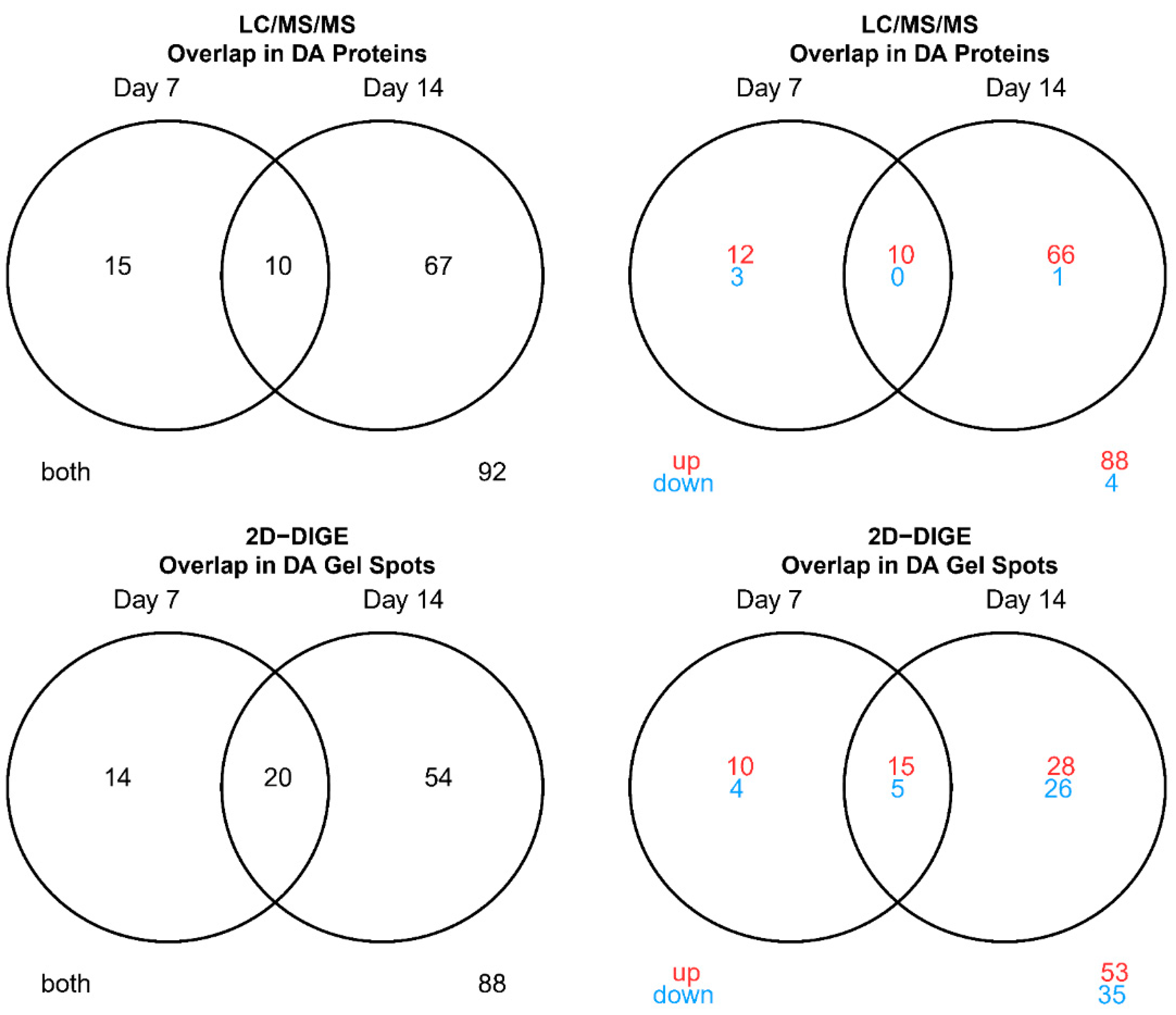
3.1. Technical Assessments and Method Comparisons
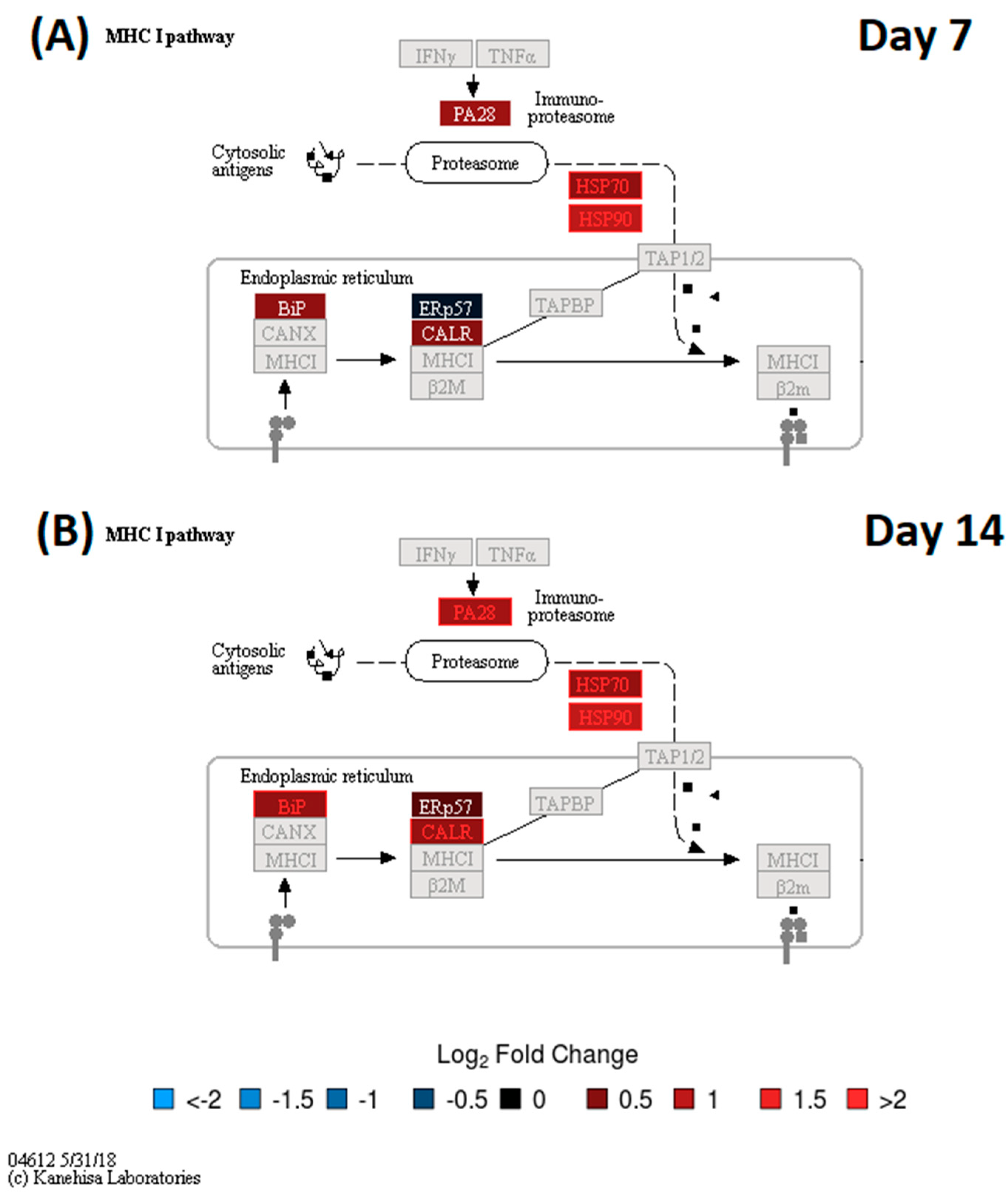
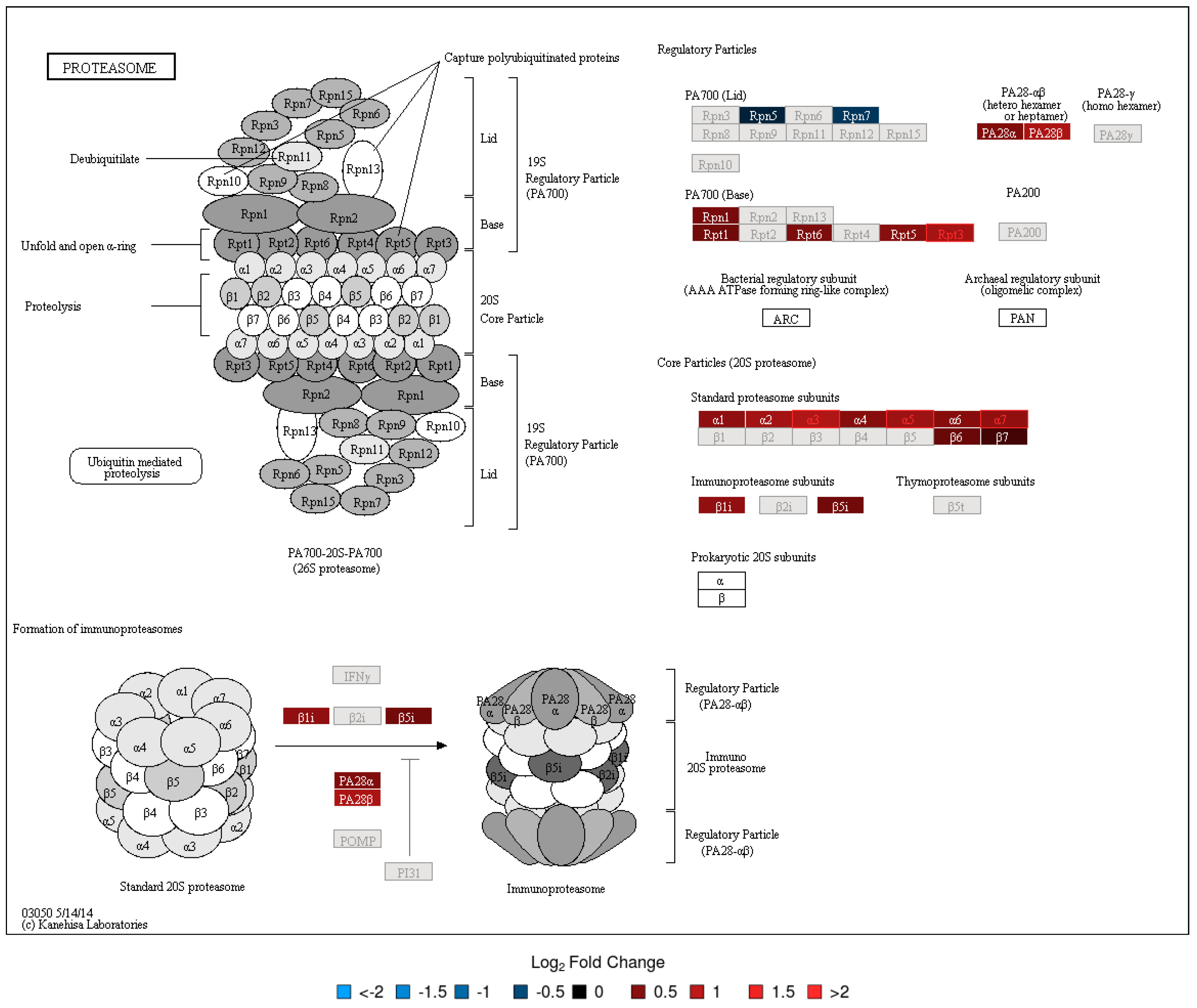
3.2. DA Proteins and Enriched Pathways
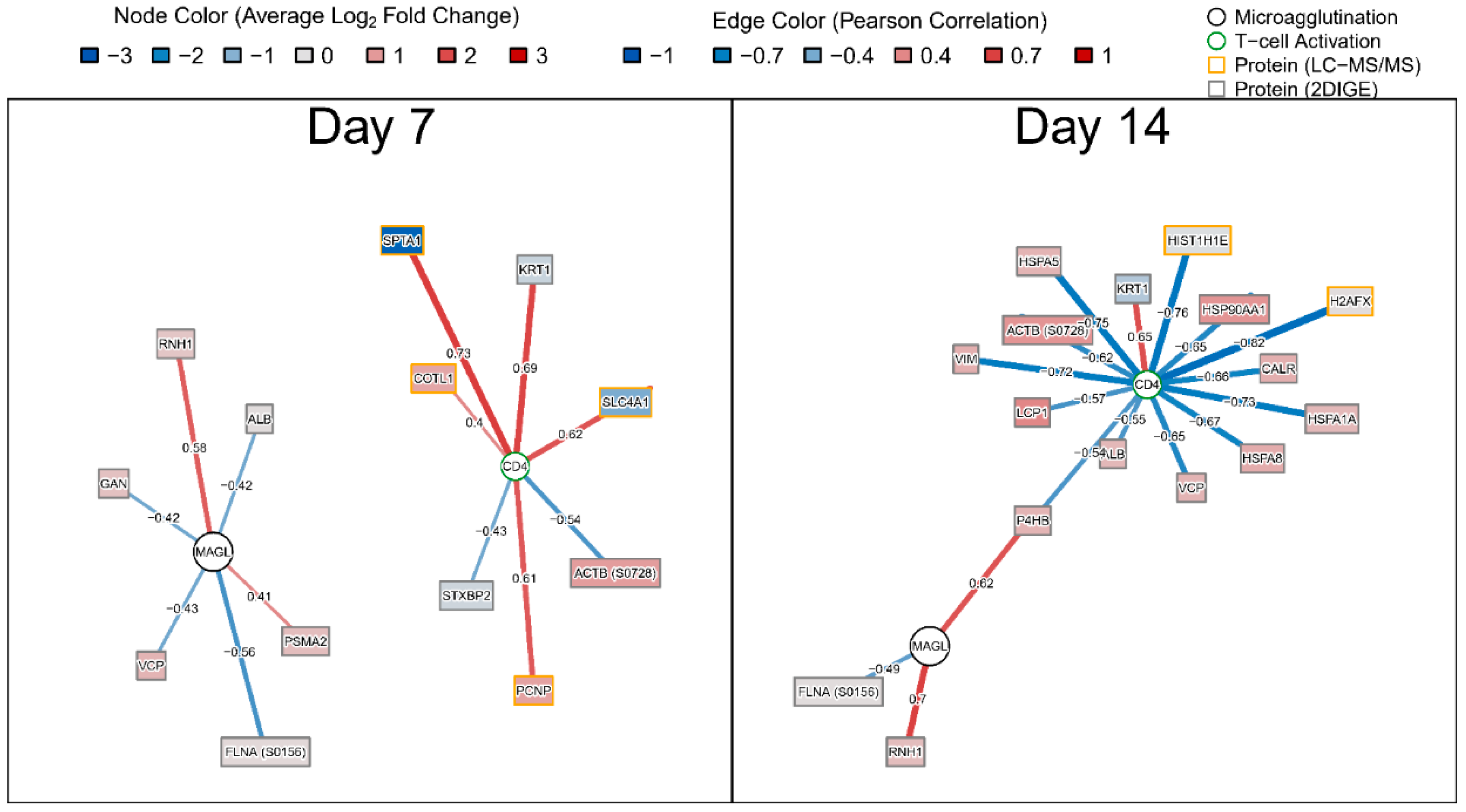
3.3. Identification of Protein Responses that Best Predicted Humoral and Cellular Responses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Francis, E. Tularemia. JAMA 1983, 250, 3216–3224. [Google Scholar] [CrossRef] [PubMed]
- McCoy, G.W.; Chapin, C.W. Further observation on a plaguelike disease of rodents with a preliminary note on the causative agent. J. Infect. Dis. 1912, 10, 61–72. [Google Scholar] [CrossRef]
- Francis, E. Deer-fly fever or Pahvant Valley Plague. A disease of man of hitherto unknown etiology. Public Health Rep. 1919, 34, 2061–2062. [Google Scholar] [CrossRef]
- Saslaw, S.; Eigelsbach, H.T.; Wilson, H.E.; Prior, J.A.; Carhart, S. Tularemia vaccine study. I. Intracutaneous challenge. Arch. Intern. Med. 1961, 107, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Saslaw, S.; Eigelsbach, H.T.; Prior, J.A.; Wilson, H.E.; Carhart, S. Tularemia vaccine study. II. Respiratory challenge. Arch. Intern. Med. 1961, 107, 702–714. [Google Scholar] [CrossRef]
- Penn, R.L. Francisella tularensis (tularemia). In Principles and Practices of Infectious Diseases, 4th ed.; Mandell, G.L., Douglas, R.G., Bennett, R.G., Bennett, J.E., Eds.; John Wiley and Sons: New York, NY, USA, 2012; pp. 2060–2067. [Google Scholar]
- Centers for Disease, C. Outbreak of tick-borne tularemia-South Dakota. MMWR Rorbid. Mortal. Week Rep. 1984, 33, 601–602. [Google Scholar]
- Dennis, D.T.; Inglesby, T.V.; Henderson, D.A.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Fine, A.D.; Friedlander, A.M.; Hauer, J.; Layton, M.; et al. Tularemia as a biological weapon: Medical and public health management. JAMA 2001, 285, 2763–2773. [Google Scholar] [CrossRef]
- Christopher, G.W.; Cieslak, T.J.; Pavlin, J.A.; Eitzen, E.M., Jr. Biological warfare. A historical perspective. JAMA 1997, 278, 412–417. [Google Scholar] [CrossRef]
- Alibek, K.; Handelman, S. Biohazard: The Chilling True Story of the Largest Covert Biological Weapons Program in the World, Told from the Inside by the Man Who Ran It; Random House: New York, NY, USA, 1999. [Google Scholar]
- Tigertt, W.D. Soviet viable Pasteurella tularensis vaccine. A review of selected articles. Bacteriol. Rev. 1962, 26, 354–373. [Google Scholar] [CrossRef]
- Pasetti, M.F.; Cuberos, L.; Horn, T.L.; Shearee, J.D.; Matthews, S.J.; House, R.V.; Sztein, M.B. An improved Francisella tularensis Live Vaccine Strain (LVS) is well tolerated and highly immunogenic when administered to rabbits in escalating doses using immunization routes. Vaccine 2008, 26, 1773–1785. [Google Scholar] [CrossRef] [Green Version]
- El Sahly, H.M.; Atmar, R.L.; Patel, S.M.; Wells, J.M.; Cate, T.; Ho, M.; Guo, K.; Pasetti, M.F.; Lewis, D.E.; Sztein, M.B.; et al. Safety, reactogenicity and immunogenicity of Francisella tularensis live vaccine strain in humans. Vaccine 2009, 27, 4905–4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan, M.J.; Stapleton, J.T.; Keitel, W.A.; Frey, S.E.; Chen, W.H.; Rouphael, N.; Edupuganti, S.; Beck, A.; Winokur, P.L.; El Sahly, H.M.; et al. Tularemia vaccine: Safety, reactogenicity, ‘‘Take” skin reactions, and antibody responses following vaccination with a new lot of the Francisella tularensis live vaccine strain—A phase 2 randomized clinical Trial. Vaccine 2017, 35, 4730–4737. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, M.S.; Rouphael, N.; Lai, L.; Kazmin, D.; Jensen, T.L.; Weiss, D.S.; Ibegbu, C.; Sztein, M.B.; Hooper, W.F.; Hill, H.; et al. Systems vaccinology for a live attenuated tularemia vaccine reveals unique transcriptional signatures that predict humoral and cellular immune responses. Vacccines 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallassie, A.C.; Link, A.J. Proteomics contributions to our understanding of vaccine and immune responses. Proteom. Clin. Appl. 2015, 9, 972–989. [Google Scholar] [CrossRef] [PubMed]
- Unlü, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef]
- Arentz, G.; Weiland, F.; Oehler, M.K.; Hoffmann, P. State of the art of 2D DIGE. Proteom. Clin. Appl. 2015, 9, 277–288. [Google Scholar] [CrossRef]
- Gant-Branum, R.L.; Kerr, T.J.; McLean, J.A. Labeling strategies in mass spectrometry-based protein quantitation. Analyst 2009, 134, 1525–1529. [Google Scholar] [CrossRef]
- Lee, J.R.; Magee, D.M.; Gaster, R.S.; LaBaer, J.; Wang, S.X. Emerging protein array technologies for proteomics. Expert Rev. Proteom. 2013, 10, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Hall, D.A.; Ptacek, J.; Snyder, M. Protein microarray technology. Mech. Ageing Dev. 2007, 128, 161–167. [Google Scholar] [CrossRef]
- Breker, M.; Schuldiner, M. The emergence of proteome-wide technologies: Systematic analysis of proteins comes of age. Nat. Rev. Mol. Cell Biol. 2014, 15, 453–464. [Google Scholar] [CrossRef]
- Maner-Smith, K.M.; Ford, D.A.; Goll, J.B.; Jensen, T.L.; Khadka, M.; Colucci, J.K.; Gelber, C.E.; Albert, C.J.; Bosinger, S.; Franke, J.D.; et al. Alterations in the Human Plasma Lipidome in Response to Tularemia Vaccination. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Crompton, J.G.; Narayanan, M.; Cuddapah, S.; Roychoudhuri, R.; Ji, Y.; Yang, W.; Patel, S.J.; Sukumar, M.; Palmer, D.C.; Peng, W.; et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell. Mol. Immunol. 2016, 13, 502–513. [Google Scholar] [CrossRef]
- Pierce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013, 324, 1242454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Set: GSE2405_0H_VS_9H_A_PHAGOCYTOPHILUM_STIM_NEUTROPHIL_DN. Available online: http://software.broadinstitute.org/gsea/msigdb/cards/GSE2405_0H_VS_9H_A_PHAGOCYTOPHILUM_STIM_NEUTROPHIL_DN (accessed on 16 March 2016).
- Borjesson, D.L.; Kobayashi, S.D.; Whitney, A.R.; Voyich, J.M.; Argue, C.M.; Deleo, F.R. Insights into pathogen immune evasion mechanisms: Anaplasma phagocytophilum fails to induce an apoptosis differentiation program in human neutrophils. J. Immunol. 2005, 174, 6364–6372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Set: GSE25123_WT_VS_PPARG_KO_MACROPHAGE_DN. Available online: http://software.broadinstitute.org/gsea/msigdb/cards/GSE25123_WT_VS_PPARG_KO_MACROPHAGE_DN (accessed on 16 March 2016).
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Gene Set: GSE37532_TREG_VS_TCONV_CD4_TCELL_FROM_LN_UP. Available online: http://software.broadinstitute.org/gsea/msigdb/cards/GSE37532_TREG_VS_TCONV_CD4_TCELL_FROM_LN_UP (accessed on 16 March 2016).
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue T reg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef]
- Gene Set: GSE37532_WT_VS_PPARG_KO_VISCERAL_ADIPOSE_TISSUE_TREG_UP. Available online: http://software.broadinstitute.org/gsea/msigdb/cards/GSE37532_WT_VS_PPARG_KO_VISCERAL_ADIPOSE_TISSUE_TREG_UP (accessed on 14 April 2016).
- Gene Set: GSE37532_TREG_VS_TCONV_PPARG_KO_CD4_TCELL_FROM_LN_DN. Available online: http://software.broadinstitute.org/gsea/msigdb/cards/GSE37532_TREG_VS_TCONV_PPARG_KO_CD4_TCELL_FROM_LN_DN (accessed on 16 July 2016).
- Kusters, B.; Schirle, M.; Mallick, P.; Aebersold, R. Scoring proteomes with proteotypic peptide probes. Nat. Rev. Mol. Cell Biol. 2005, 6, 577–583. [Google Scholar] [CrossRef]
- Lim, H.; Eng, J.; Yates, J.R., 3rd; Tollaksen, S.L.; Giometti, C.S.; Holden, J.F.; Adams, M.W.W.; Reich, C.I.; Olsen, G.J.; Hays, L.G. Identification of 2D-gel proteins: A comparison of MALDI/TOF peptide mass mapping to μ LC-ESI tandem mass spectrometry. J. Am. Soc. Mass Spectrum. 2003, 14, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.W.; Wang, G.; Baek, S.J.; Shen, R.F. Comparative study of three proteomic quantitative methods, DIGE, cICAT, and iTRAQ, using 2D gel-or LC-MALDI TOF/TOF. J. Proteome Res. 2006, 5, 651–658. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Ament, Z.; West, J.A.; Stanley, E.; Ashmore, T.; Roberts, L.D.; Wright, J.; Nicholls, A.W.; Griffin, J.L. PPAR-pan activation induces hepatic oxidative stress and lipidomic remodeling. Free Radic. Biol. Med. 2016, 95, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroker, A.J.; Bruning, J.B. Review of the structure and dynamic mechanisms of PPARγ partial agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guimarães, G.; Gomes, M.T.R.; Campos, P.C.; Marinho, F.V.; de Assis, N.R.G.; Silveira, T.N.; Oliveira, S.C. Immunoproteasome Subunits Are Required for CD8+ T Cell Function and Host Resistance to Brucella abortus Infection in Mice. Infect. Immunity 2018, 86, e00615-17. [Google Scholar] [CrossRef] [Green Version]
- Galassie, A.C.; Goll, J.B.; Samir, P.; Jensen, T.L.; Hoek, K.L.; Howard, L.M.; Allos, T.M.; Niu, X.; Gordy, L.E.; Creech, C.B.; et al. Proteomics show antigen presentation processes in human immune cells after AS03-H5N1 vaccination. Proteomics 2017, 17, 1600453. [Google Scholar] [CrossRef]
- Kim, J.; Shapiro, M.J.; Bamidele, A.O.; Gurel, P.; Thapa, P.; Higgs, H.N.; Hedin, K.E.; Shapiro, V.S.; Billadeau, D.D. Coactosin-Like 1 Antagonizes Cofilin to Promote Lamellipodial Protrusion at the Immune Synapse. PLoS ONE 2014, 9, e85090. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | Gene Name | Protein Description | 50% Protein Cluster ID (Gene Name) | LC-MS/MSTime Point | 2D-DIGE/MS Time Point | LC/MS/MS Log2 Fold Change | 2D-DIGE Log2 Fold Change |
|---|---|---|---|---|---|---|---|
| P33176 | KIF5B | Kinesin-1 heavy chain | Q12840 (KIF5A) | Day 7 | Day 7 | 1.11 | 0.33 |
| P33176 | KIF5B | Kinesin-1 heavy chain | Q12840 (KIF5A) | Day 7 | Day 14 | 1.11 | 0.35 |
| P07900 | HSP90AA1 | Heat shock protein HSP 90-alpha | P07900 (HSP90AA1) | Day 14 | Day 7 | 1.12 | 1.00 |
| P60174 | TPI1 | Triosephosphate isomerase | P60174 (TPI1) | Day 14 | Day 7 | 1.12 | 0.58 |
| P07900 | HSP90AA1 | Heat shock protein HSP 90-alpha | P07900 (HSP90AA1) | Day 14 | Day 14 | 1.12 | 1.02 |
| P52907 | CAPZA1 | F-actin-capping protein subunit alpha-1 | P52907 (CAPZA1) | Day 14 | Day 14 | 0.56 | 0.58 |
| Assay | Day | Pathway | Pathway Genes # | Significant Genes (%) [Protein Families] | Genes Up (%) | Genes Down (%) | Jaccard Similarity Coefficient | p | FDR-Adjusted P |
|---|---|---|---|---|---|---|---|---|---|
| LC-MS/MS | Day 7 | Ribosome | 127 | 5 (3.9) [5] | 5 (3.9) | 0 (0) | 0.036 | <0.0001 | 0.003 |
| Day 14 | Ribosome | 127 | 13 (10.2) [14] | 13 (10.2) | 0 (0) | 0.0798 | <0.0001 | <0.0001 | |
| Proteasome | 44 | 4 (9.1) [4] | 4 (9.1) | 0 (0) | 0.0449 | 0.0003 | 0.0473 | ||
| 2-DIGE/MS | Day 7 | Pathogenic Escherichia coli infection | 54 | 3 (5.6) [3] | 3 (5.6) | 0 (0) | 0.2273 | 0.0001 | 0.0287 |
| Antigen processing and presentation | 64 | 3 (4.7) [2] | 3 (4.7) | 0 (0) | 0.046 | 0.0002 | 0.0287 | ||
| Estrogen signaling pathway | 97 | 3 (3.1) [2] | 3 (3.1) | 0 (0) | 0.0333 | 0.0006 | 0.0653 | ||
| Day 14 | Protein processing in endoplasmic reticulum | 159 | 8 (5) [7] | 8 (5) | 0 (0) | 0.0481 | <0.0001 | <0.0001 | |
| Antigen processing and presentation | 64 | 5 (7.8) [4] | 5 (7.8) | 0 (0) | 0.0745 | <0.0001 | 0.0002 | ||
| Pathogenic Escherichia coli infection | 54 | 4 (7.4) [4] | 4 (7.4) | 0 (0) | 0.2133 | <0.0001 | 0.0023 | ||
| Estrogen signaling pathway | 97 | 4 (4.1) [3] | 4 (4.1) | 0 (0) | 0.0388 | 0.0002 | 0.0175 | ||
| Legionellosis | 48 | 3 (6.2) [2] | 3 (6.2) | 0 (0) | 0.0366 | 0.0005 | 0.0279 | ||
| Phagosome | 142 | 4 (2.8) [4] | 4 (2.8) | 0 (0) | 0.0982 | 0.001 | 0.0498 |
| LC-MS/MS | 2D-DIGE/MS | ||||||
|---|---|---|---|---|---|---|---|
| Category Name | Category Genes | Sig. Genes (%) [Protein Fam.] | Jaccard Similarity Coefficient | FDR | Sig. Genes (%) [Protein Fam] | Jaccard Similarity Coefficient | FDR |
| Day 7 | |||||||
| GSE41978 KLRG1 HIGH VS LOW EFFECTOR CD8 TCELL DN | 186 | 5 (2.7) [5] | 0.0244 | 0.0101 | 3 (1.6) [3] | 0.0234 | 0.0543 |
| GSE41978 ID2 KO VS ID2 KO AND BIM KO KLRG1 LOW EFFECTOR CD8 TCELL DN | 190 | 5 (2.6) [5] | 0.0239 | 0.0101 | 5 (2.6) [5] | 0.0324 | 0.0006 |
| GSE2405 0H VS 9H A PHAGOCYTOPHILUM STIM NEUTROPHIL DN | 192 | 5 (2.6) [5] | 0.0237 | 0.0101 | 5 (2.6) [5] | 0.0321 | 0.0006 |
| GSE3982 EOSINOPHIL VS MAST CELL DN | 186 | 4 (2.2) [4] | 0.0194 | 0.0999 | 3 (1.6) [3] | 0.0186 | 0.0543 |
| GSE2405 0H VS 24H A PHAGOCYTOPHILUM STIM NEUTROPHIL UP | 193 | 4 (2.1) [4] | 0.0188 | 0.0999 | 3 (1.6) [3] | 0.0226 | 0.0543 |
| Day 14 | |||||||
| GSE2405 0H VS 9H A PHAGOCYTOPHILUM STIM NEUTROPHIL DN | 192 | 20 (10.4) [20] | 0.0813 | <0.0001 | 8 (4.2) [8] | 0.0485 | <0.0001 |
| GSE2405 0H VS 24H A PHAGOCYTOPHILUM STIM NEUTROPHIL UP | 193 | 17 (8.8) [17] | 0.068 | <0.0001 | 4 (2.1) [4] | 0.0302 | 0.029 |
| GSE41978 ID2 KO VS ID2 KO AND BIM KO KLRG1 LOW EFFECTOR CD8 TCELL DN | 190 | 12 (6.3) [12] | 0.0476 | <0.0001 | 5 (2.6) [5] | 0.0306 | 0.0045 |
| GSE22886 NAIVE CD8 TCELL VS DC DN | 187 | 5 (2.7) [5] | 0.0195 | 0.0598 | 4 (2.1) [4] | 0.0264 | 0.029 |
| GSE29618 BCELL VS MDC DAY7 FLU VACCINE DN | 191 | 5 (2.6) [5] | 0.0192 | 0.0598 | 5 (2.6) [5] | 0.0304 | 0.0045 |
| GSE23114 PERITONEAL CAVITY B1A BCELL VS SPLEEN BCELL DN | 194 | 5 (2.6) [5] | 0.019 | 0.0601 | 4 (2.1) [4] | 0.0213 | 0.029 |
| GSE2405_0H_VS_24H_A_PHAGOCYTOPHILUM_STIM_NEUTROPHIL_UP | |||
| Assay | Gene | UniProt ID | Day 14 Fold Change |
| 2D DIGE/MS | ACTB;ACTG1 | P60709 | 2.271;2.004 |
| TUBA1A;TUBA1B;TUBA1C;TUBA3D;TUBA3E;TUBA8 | P68363 | 2.046 | |
| HSPA8 | P11142 | 1.629 | |
| HSPA5 | P11021 | 1.522 | |
| LC MS/MS | NPM1 | P06748 | 2.02 |
| TKT | P29401 | 1.784 | |
| RPS21 | P63220 | 1.702 | |
| RPL30 | P62888 | 1.638 | |
| RPL4 | P36578 | 1.602 | |
| RPL5 | P46777 | 1.559 | |
| RPL7 | P18124 | 1.555 | |
| RPL22 | P35268 | 1.526 | |
| RPS29 | P62273 | 1.503 | |
| RPL3 | P39023 | 1.495 | |
| RPL17 | P18621 | 1.449 | |
| EEF1B2 | P24534 | 1.433 | |
| RPS18 | P62269 | 1.428 | |
| RPS27 | P42677 | 1.349 | |
| RPL6 | Q02878 | 1.327 | |
| EIF3I | Q13347 | 1.309 | |
| BRK1 | Q8WUW1 | 1.271 | |
| GSE2405_0H_VS_9H_A_PHAGOCYTOPHILUM_STIM_NEUTROPHIL_DN | |||
| Assay | Gene | UniProt ID | Day 14 Fold Change |
| 2D DIGE/MS | ACTB;ACTG1 | P60709 | 2.271;2.004 |
| TUBA1A;TUBA1B;TUBA1C;TUBA3D;TUBA3E;TUBA8 | P68363 | 2.046 | |
| HSP90AA1;HSP90AB1 | P07900 | 2.036 | |
| TUBB;TUBB2A;TUBB2B;TUBB3;TUBB4A;TUBB4B | P07437 | 1.857 | |
| YWHAZ | P63104 | 1.848 | |
| CAPZB;CEP128;PSME1 | P47756 | 1.674 | |
| HSPA8 | P11142 | 1.629 | |
| VCP | P55072 | 1.498 | |
| LC MS/MS | HSP90AA1 | P07900 | 2.183 |
| NPM1 | P06748 | 2.02 | |
| EIF4A1 | P60842 | 1.838 | |
| TKT | P29401 | 1.784 | |
| HMGB1 | P09429 | 1.75 | |
| RPS21 | P63220 | 1.702 | |
| RPL30 | P62888 | 1.638 | |
| RPL4 | P36578 | 1.602 | |
| RPL5 | P46777 | 1.559 | |
| RPL7 | P18124 | 1.555 | |
| CHTOP | Q9Y3Y2 | 1.539 | |
| RPL22 | P35268 | 1.526 | |
| RPS29 | P62273 | 1.503 | |
| RPL3 | P39023 | 1.495 | |
| RPL17 | P18621 | 1.449 | |
| EEF1B2 | P24534 | 1.433 | |
| RPS18 | P62269 | 1.428 | |
| RPS27 | P42677 | 1.349 | |
| RPL6 | Q02878 | 1.327 | |
| EIF3I | Q13347 | 1.309 | |
| GSE25123_WT_VS_PPARG_KO_MACROPHAGE_DN | |||
| Assay | Gene | UniProt ID | Day 14 Fold Change |
| 2D DIGE/MS | ARHGDIB | P52566 | 1.504 |
| LC MS/MS | SYNCRIP | O60506 | 2.136 |
| SAMHD1 | Q9Y3Z3 | 2.118 | |
| PSMA4 | P25789 | 1.814 | |
| PTBP1 | P26599 | 1.635 | |
| PSMA5 | P28066 | 1.595 | |
| RPL5 | P46777 | 1.559 | |
| RPL7 | P18124 | 1.555 | |
| EEF1B2 | P24534 | 1.433 | |
| SNX3 | O60493 | 1.247 | |
| GSE37532_TREG_VS_TCONV_CD4_TCELL_FROM_LN_UP | |||
| 2D DIGE/MS | HSP90B1 | P14625 | 1.636 |
| LC MS/MS | ATIC | P31939 | 2.222 |
| NOP56 | O00567 | 2.169 | |
| CCT3 | P49368 | 2.018 | |
| HDGF | P51858 | 1.801 | |
| EEF1D | P29692 | 1.578 | |
| NOP58 | Q9Y2X3 | 1.525 | |
| SMAP | O00193 | 1.452 | |
| GSE37532_TREG_VS_TCONV_PPARG_KO_CD4_TCELL_FROM_LN_DN | |||
| 2D DIGE/MS | LCP1 | P13796 | 2.293 |
| CALR | P27797 | 1.558 | |
| HSPA5 | P11021 | 1.522 | |
| P4HB | P07237 | 1.505 | |
| GSE37532_WT_VS_PPARG_KO_VISCERAL_ADIPOSE_TISSUE_TREG_UP | |||
| 2D DIGE/MS | YWHAZ | P63104 | 1.848 |
| LC MS/MS | HDGF | P51858 | 1.801 |
| HMGB2 | P26583 | 1.757 | |
| TCEA1 | P23193 | 1.661 | |
| PPP6C | O00743 | 1.638 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-H.; Duong, D.M.; Goll, J.B.; Wood, D.C.; Jensen, T.L.; Yin, L.; Gelber, C.E.; Seyfried, N.T.; Anderson, E.; Natrajan, M.S.; et al. Proteomic Analysis of Human Immune Responses to Live-Attenuated Tularemia Vaccine. Vaccines 2020, 8, 413. https://doi.org/10.3390/vaccines8030413
Chang Y-H, Duong DM, Goll JB, Wood DC, Jensen TL, Yin L, Gelber CE, Seyfried NT, Anderson E, Natrajan MS, et al. Proteomic Analysis of Human Immune Responses to Live-Attenuated Tularemia Vaccine. Vaccines. 2020; 8(3):413. https://doi.org/10.3390/vaccines8030413
Chicago/Turabian StyleChang, Yie-Hwa, Duc M. Duong, Johannes B. Goll, David C. Wood, Travis L. Jensen, Luming Yin, Casey E. Gelber, Nicholas T. Seyfried, Evan Anderson, Muktha S. Natrajan, and et al. 2020. "Proteomic Analysis of Human Immune Responses to Live-Attenuated Tularemia Vaccine" Vaccines 8, no. 3: 413. https://doi.org/10.3390/vaccines8030413
APA StyleChang, Y.-H., Duong, D. M., Goll, J. B., Wood, D. C., Jensen, T. L., Yin, L., Gelber, C. E., Seyfried, N. T., Anderson, E., Natrajan, M. S., Rouphael, N., Johnson, R. A., Sanz, P., Mulligan, M. J., & Hoft, D. F. (2020). Proteomic Analysis of Human Immune Responses to Live-Attenuated Tularemia Vaccine. Vaccines, 8(3), 413. https://doi.org/10.3390/vaccines8030413