Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach

 ,
,  ,
,  ,
, 
 ,
, 
 and
and 
Abstract
:1. Introduction
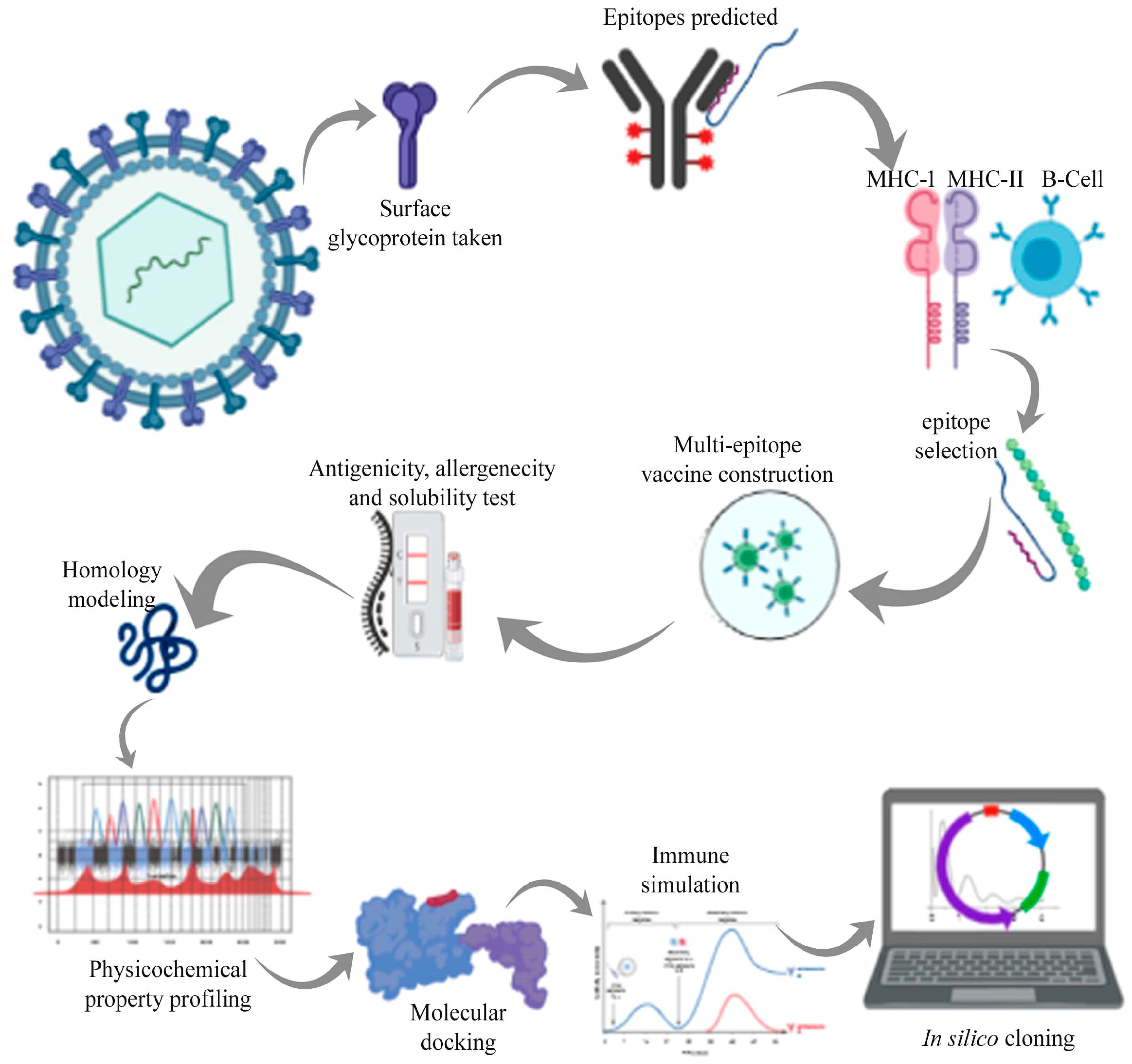
2. Materials and Methods
2.1. Protein Sequence Retrieval
2.2. Epitope Prediction
2.3. Epitopes Selection and Vaccine Construction
2.4. Determination of Antigenicity, Allergenicity, Toxicity, and Solubility
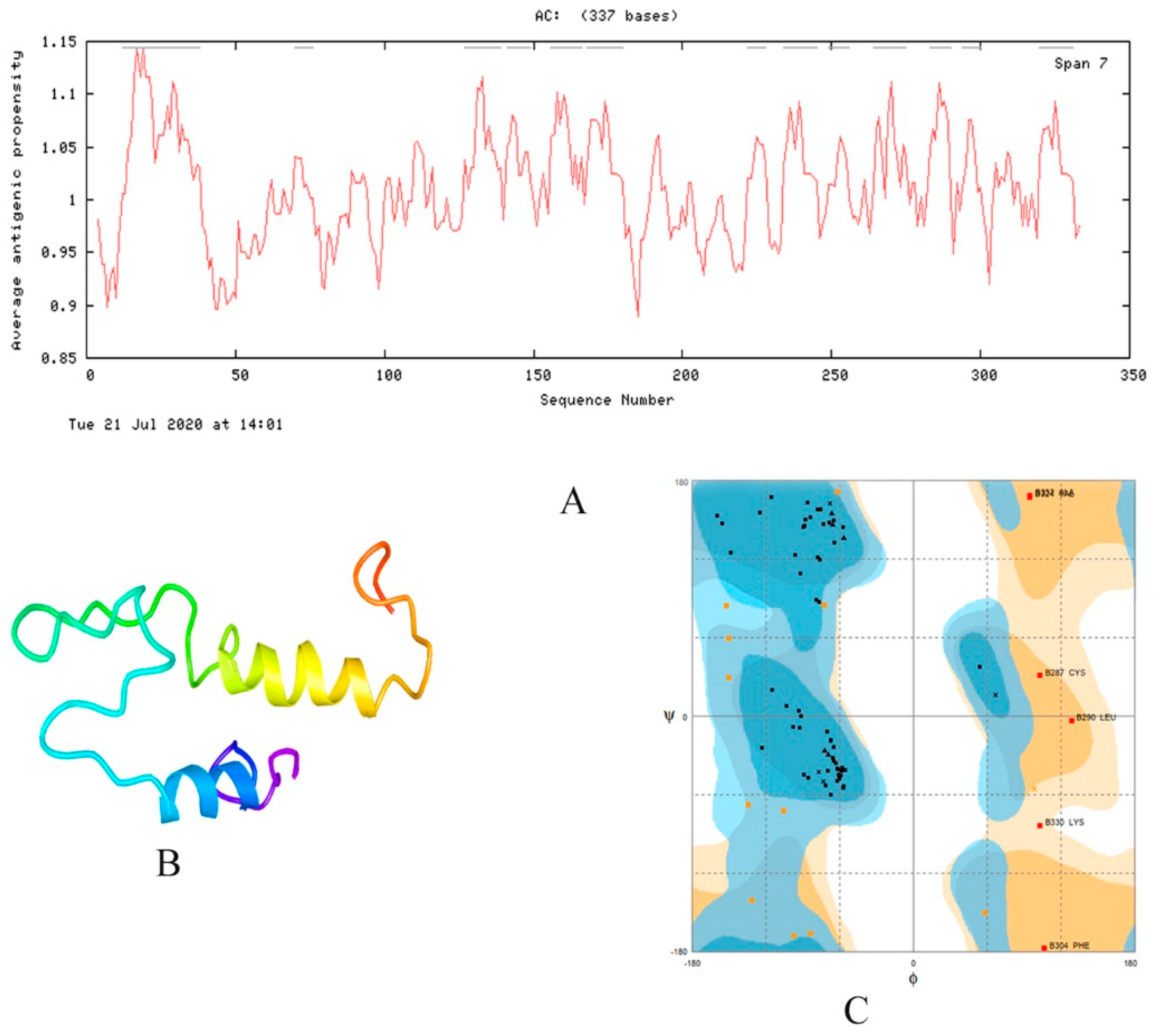
2.5. Physiochemical Properties
2.6. Homology Modeling and Model Validation
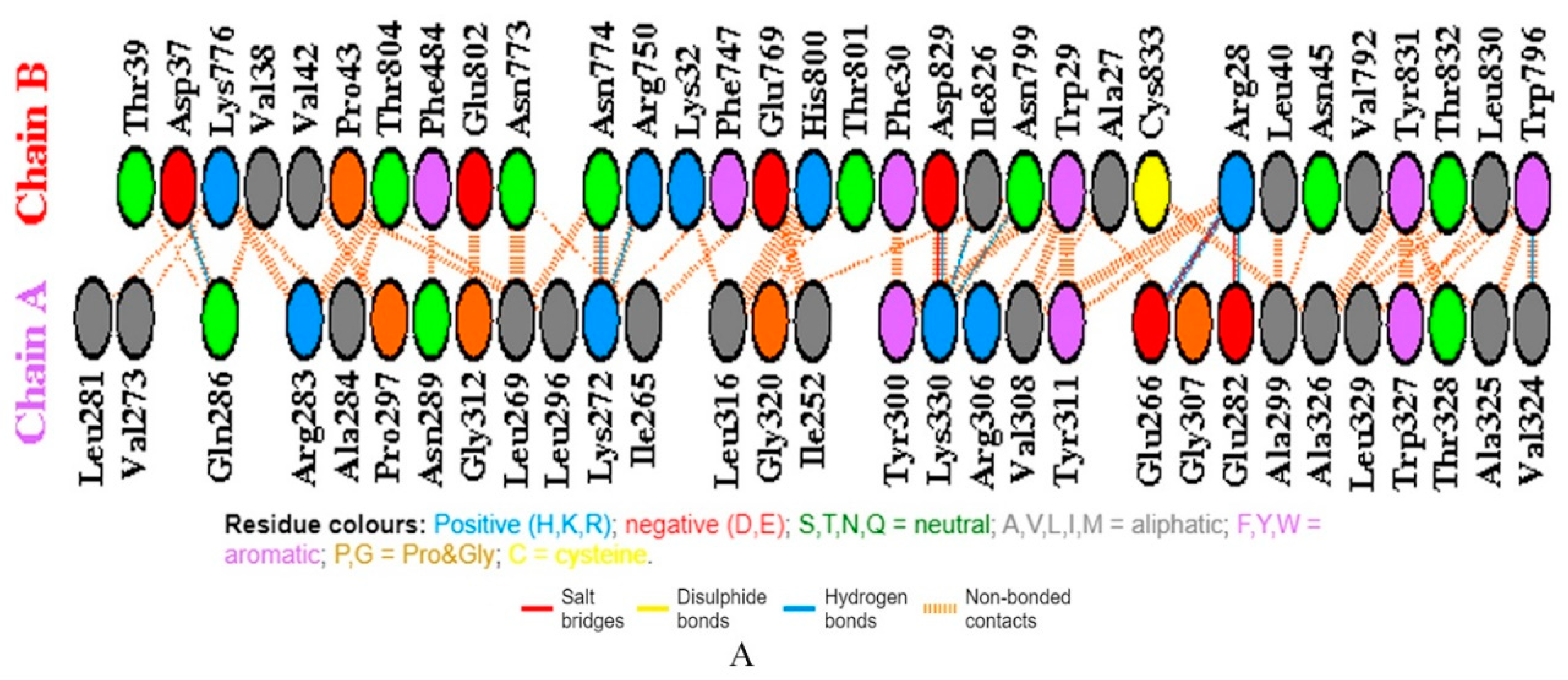
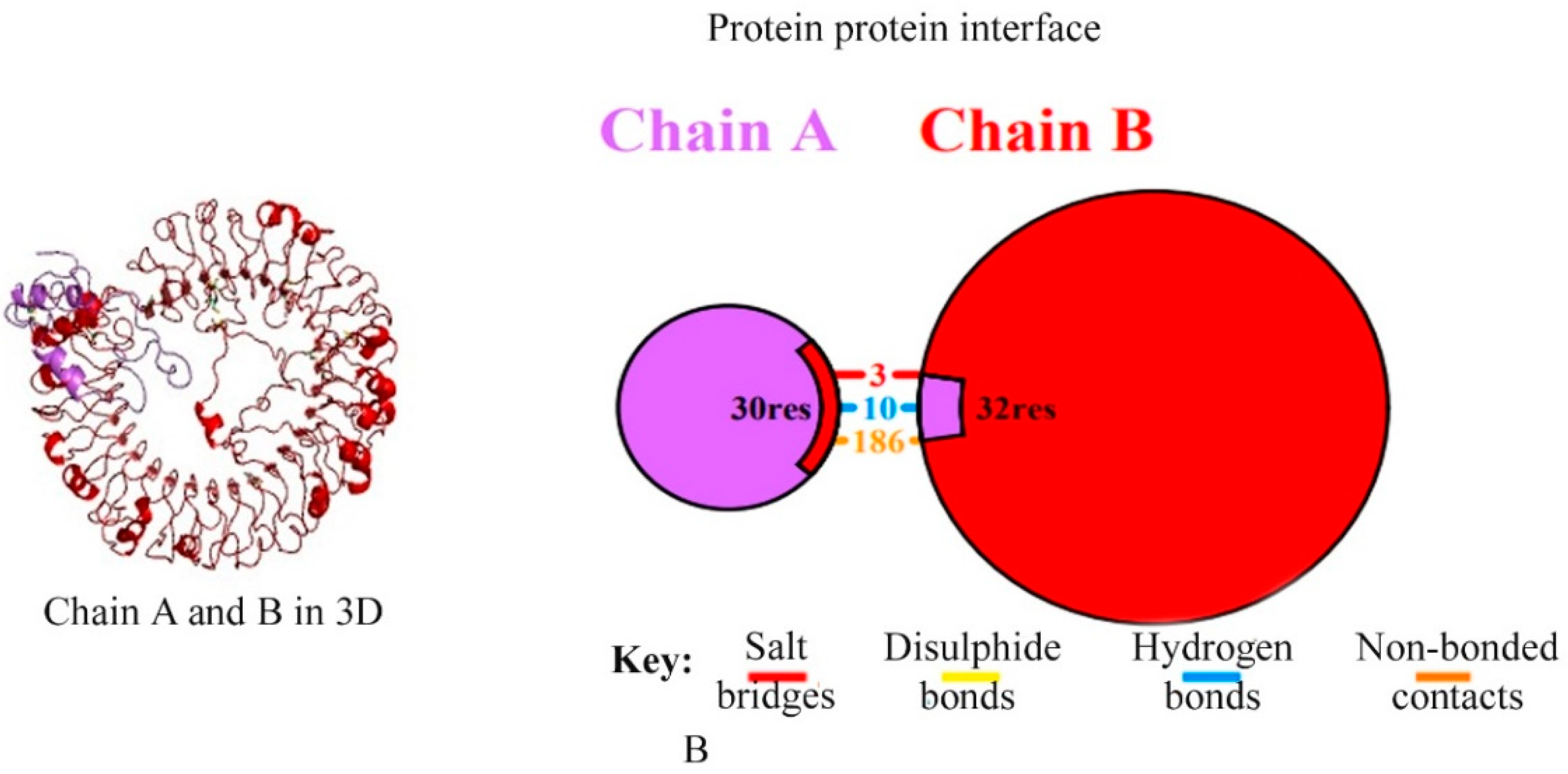
2.7. Molecular Docking and MD Simulation
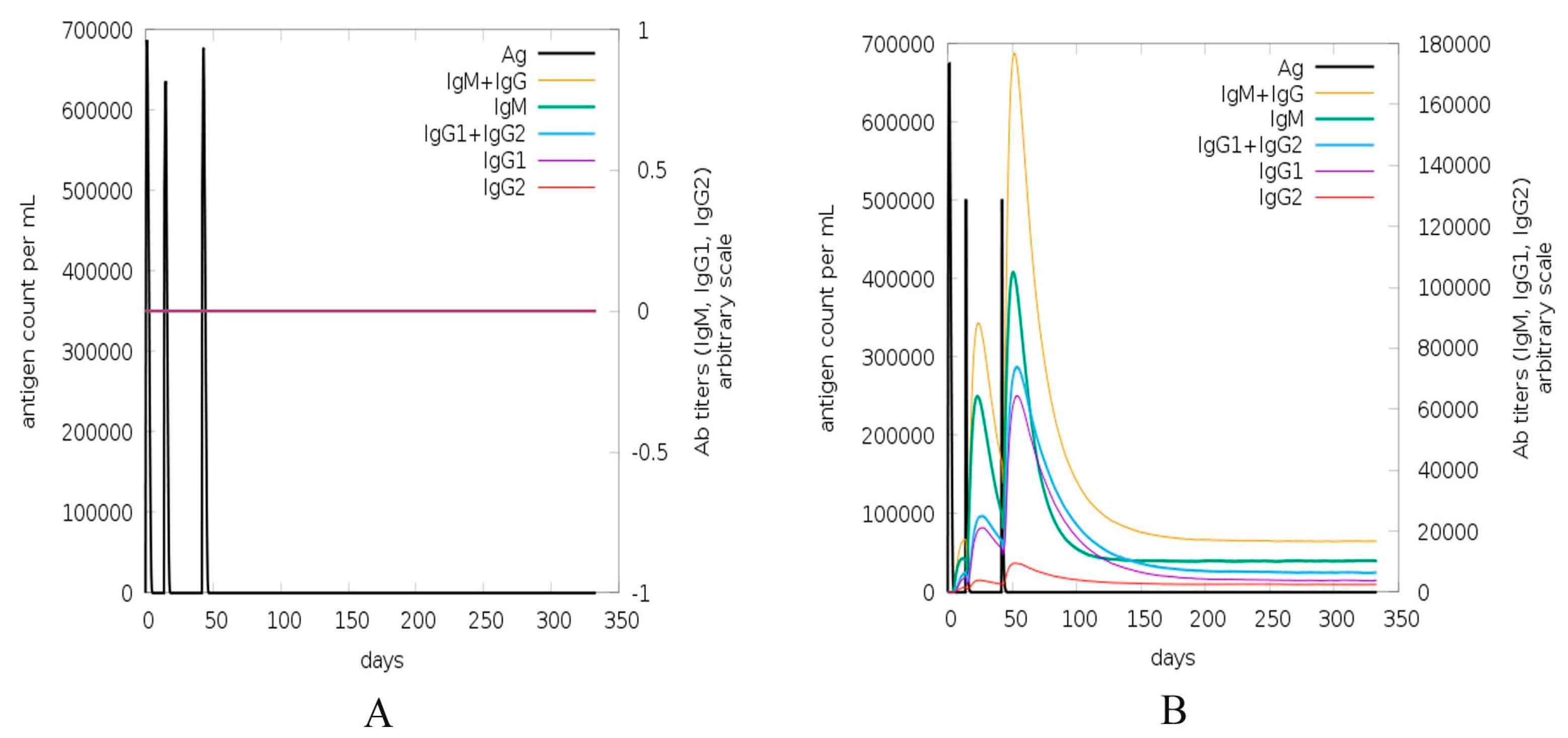
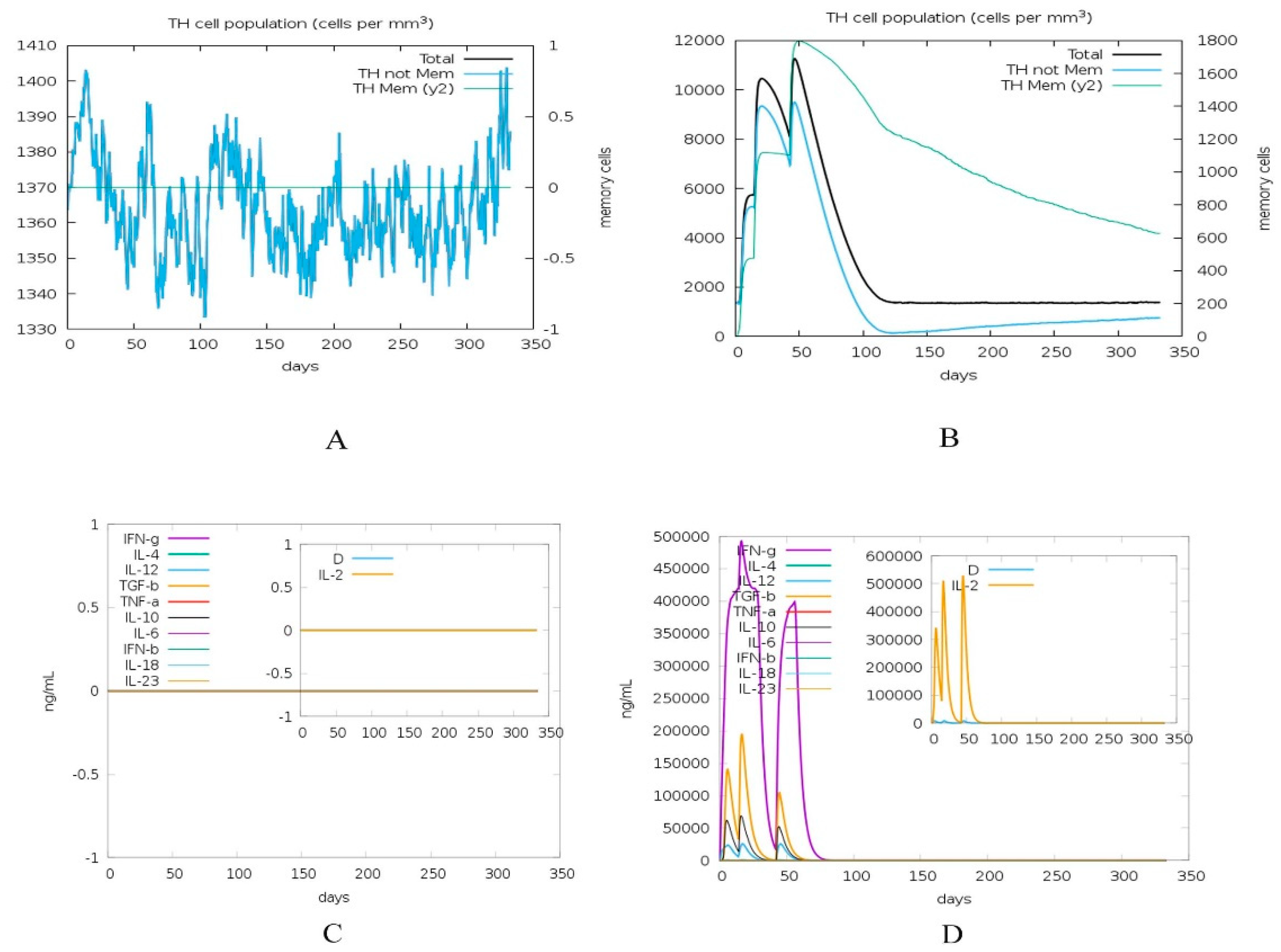
2.8. Immune Simulation of Vaccine Construct
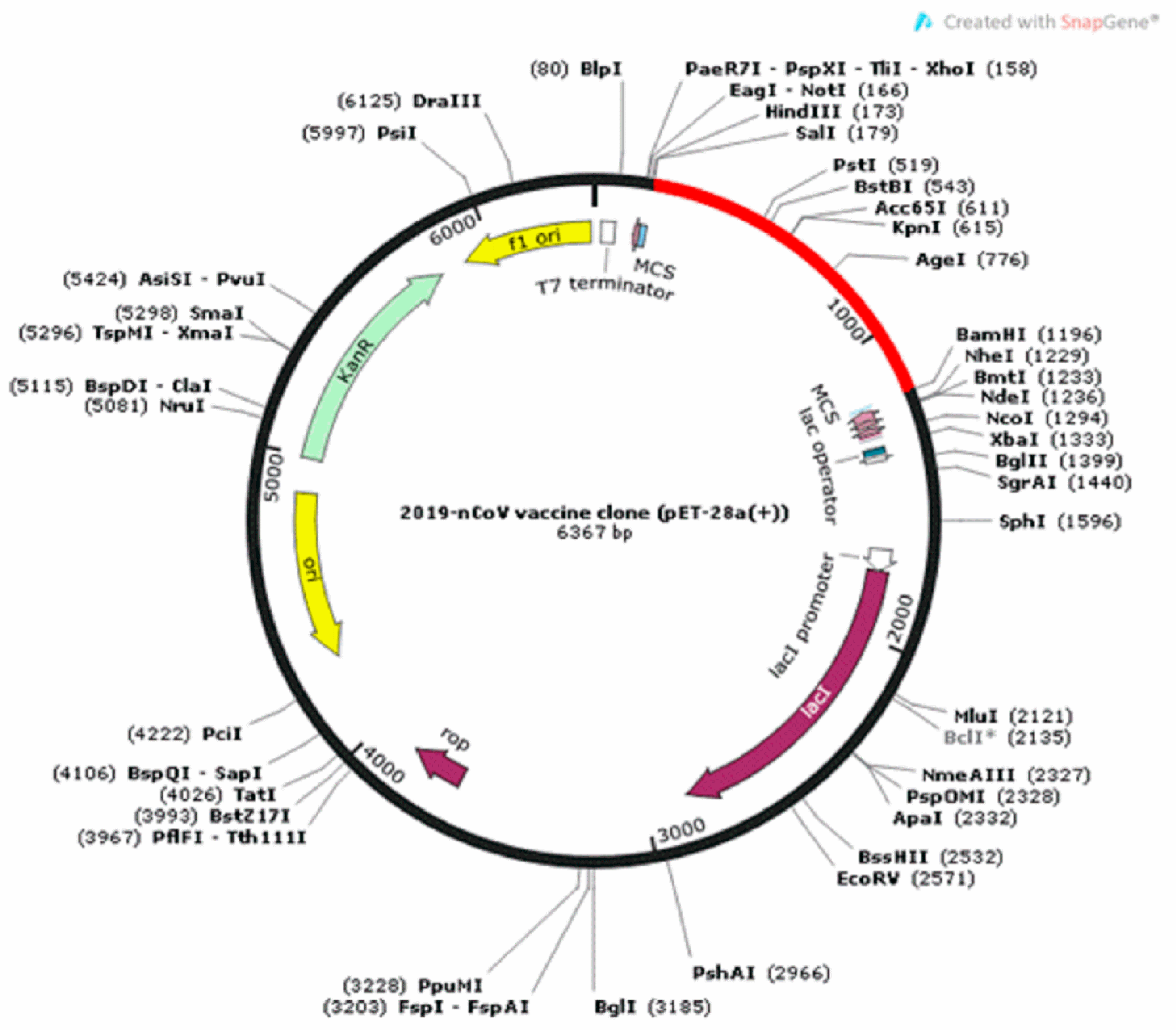
2.9. Codon Optimization and In Silico Cloning
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Articles Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 6736, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Alejandra Tortorici, M.; Walls, A.C.; Lang, Y.; Wang, C.; Li, Z.; Koerhuis, D.; Boons, G.J.; Bosch, B.J.; Rey, F.A.; de Groot, R.J.; et al. Structural basis for human coronavirus attachment to sialic acid receptors. Nat. Struct. Mol. Biol. 2019, 26, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining “host jump” of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Haagmans, B.L.; Al Dhahiry, S.H.S.; Reusken, C.B.E.M.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2014, 14, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Zheng, M.; Song, L. Novel antibody epitopes dominate the antigenicity of spike glycoprotein in SARS-CoV-2 compared to SARS-CoV. Cell. Mol. Immunol. 2020, 17, 536–538. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L. Virtual Screening of Natural Products Against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef]
- Li, Z.; Tomlinson, A.C.A.; Wong, A.H.M.; Zhou, D.; Desforges, M.; Talbot, P.J.; Benlekbir, S.; Rubinstein, J.L.; Rini, J.M. The human coronavirus HCoV-229E S-protein structure and receptor binding. Elife 2019, 8, 1–22. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shey, R.A.; Ghogomu, S.M.; Esoh, K.K.; Nebangwa, N.D.; Shintouo, C.M.; Nongley, N.F.; Asa, B.F.; Ngale, F.N.; Vanhamme, L.; Souopgui, J. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Sidney, J.; Dow, C.; Mothé, B.; Sette, A.; Peters, B. A Systematic Assessment of MHC Class II Peptide Binding Predictions and Evaluation of a Consensus Approach. PLoS Comput. Biol. 2008, 4, e1000048. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E.; et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–W530. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.; Ajmal, A.; Ali, F.; Rastrelli, L. Core proteome mediated therapeutic target mining and multi-epitope vaccine design for Helicobacter pylori. Genomics 2020. [Google Scholar] [CrossRef]
- El-manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef]
- Ahmad, T.A.; Eweida, A.E.; Sheweita, S.A. B-cell epitope mapping for the design of vaccines and effective diagnostics. Trials Vaccinol. 2016, 5, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Rosa, D.S.; Tzelepis, F.; Cunha, M.G.; Soares, I.S.; Rodrigues, M.M. The pan HLA DR-binding epitope improves adjuvant-assisted immunization with a recombinant protein containing a malaria vaccine candidate. Immunol. Lett. 2004, 92, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Jung, D.; Jeong, S.K.; Lee, C.M.; Noh, K.T.; Heo, D.R.; Shin, Y.K.; Yun, C.H.; Koh, W.J.; Akira, S.; Whang, J.; et al. Enhanced efficacy of therapeutic cancer vaccines produced by Co-treatment with mycobacterium tuberculosis heparin-binding hemagglutinin, a novel TLR4 agonist. Cancer Res. 2011, 71, 2858–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferris, L.K.; Mburu, Y.K.; Mathers, A.R.; Fluharty, E.R.; Larregina, A.T.; Ferris, R.L.; Falo, L.D. Human beta-defensin 3 induces maturation of human langerhans cell-like dendritic cells: An antimicrobial peptide that functions as an endogenous adjuvant. J. Invest. Dermatol. 2013, 133, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.J.; Jang, G.Y.; Kim, Y.S.; Park, J.H.; Lee, S.E.; Vo, M.C.; Lee, J.J.; Han, H.D.; Jung, I.D.; Kang, T.H.; et al. A novel TLR4 binding protein, 40S ribosomal protein S3, has potential utility as an adjuvant in a dendritic cell-based vaccine. J. Immunother. Cancer 2019, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mizel, S.B.; Bates, J.T. Flagellin as an Adjuvant: Cellular Mechanisms and Potential. J. Immunol. 2010, 185, 5677–5682. [Google Scholar] [CrossRef] [Green Version]
- Gnjatic, S.; Sawhney, N.B.; Bhardwaj, N. Toll-Like Receptor Agonists. Cancer J. 2010, 16, 382–391. [Google Scholar] [CrossRef] [Green Version]
- Nezafat, N.; Ghasemi, Y.; Javadi, G.; Khoshnoud, M.J.; Omidinia, E. A novel multi-epitope peptide vaccine against cancer: An in silico approach. J. Theor. Biol. 2014, 349, 121–134. [Google Scholar] [CrossRef]
- Solanki, V.; Tiwari, V. Subtractive proteomics to identify novel drug targets and reverse vaccinology for the development of chimeric vaccine against Acinetobacter baumannii. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.; Flower, D.R. Bioinformatic approach for identifying parasite and fungal candidate subunit vaccines. Open Vaccine J. 2008, 1, 4. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Modelos, C. Trabajo práctico N 13. Varianzas en función de variable independiente categórica. Nat. Protoc. 2016, 10, 845–858. [Google Scholar]
- Rahman, N.; Muhammad, I.; Nayab, G.E.; Khan, H.; Aschner, M.; Filosa, R.; Daglia, M. Molecular Docking of Isolated Alkaloids for Possible α-Glucosidase Inhibition. Biomolecules 2019, 9, 544. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, I.; Rahman, N.; Nayab, G.E.; Niaz, S.; Shah, M.; Afridi, S.G.; Khan, H.; Daglia, M.; Capanoglu, E. The Molecular Docking of Flavonoids Isolated from Daucus carota as a Dual Inhibitor of MDM2 and MDMX. Recent Pat. Anticancer. Drug Discov. 2020, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mashiach, E.; Schneidman-Duhovny, D.; Andrusier, N.; Nussinov, R.; Wolfson, H.J. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008, 36, 229–232. [Google Scholar] [CrossRef]
- Kaba, S.A.; Karch, C.P.; Seth, L.; Ferlez, K.M.B.; Storme, C.K.; Pesavento, D.M.; Laughlin, P.Y.; Bergmann-Leitner, E.S.; Burkhard, P.; Lanar, D.E. Self-assembling protein nanoparticles with built-in flagellin domains increases protective efficacy of a Plasmodium falciparum based vaccine. Vaccine 2018, 36, 906–914. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [Green Version]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, 526–531. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F.; Diego, S.; Jolla, L. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; Volume 112, pp. 531–552. [Google Scholar]
- Mohan, R.; Venugopal, S. Computational structural and functional analysis of hypothetical proteins of Staphylococcus aureus. Bioinformation 2012, 8, 722. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, S.; Jernigan, R.L. Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 1996, 256, 623–644. [Google Scholar] [CrossRef]
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 1–7. [Google Scholar] [CrossRef]
- Ghaffari-Nazari, H.; Tavakkol-Afshari, J.; Jaafari, M.R.; Tahaghoghi-Hajghorbani, S.; Masoumi, E.; Jalali, S.A. Improving multi-epitope long peptide vaccine potency by using a strategy that enhances CD4+ T Help in BALB/c mice. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, W.; Guo, J.; Zhao, G.; Sun, S.; Yu, H.; Guo, Y.; Li, J.; Jin, X.; Du, L.; et al. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccines Immunother. 2015, 11, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swati, S.; Ashok, S. Bioinformatics approaches for structural and functional analysis of proteins in secondary metabolism in Withania somnifera. Mol. Biol. Rep. 2014. [Google Scholar] [CrossRef]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: A novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Olejnik, J.; Hume, A.J.; Mühlberger, E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [Green Version]
- Gorden, K.B.; Gorski, K.S.; Gibson, S.J.; Kedl, R.M.; Kieper, W.C.; Qiu, X.; Tomai, M.A.; Alkan, S.S.; Vasilakos, J.P. Synthetic TLR Agonists Reveal Functional Differences between Human TLR7 and TLR8. J. Immunol. 2005, 174, 1259–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, N.; Bruhn, K.W.; Nguyen, B.D.; Prins, R.; Lin, J.W.; Liau, L.M.; Miller, J.F. The TLR7 Agonist Imiquimod Enhances the Anti-Melanoma Effects of a Recombinant Listeria monocytogenes Vaccine. J. Immunol. 2005, 175, 1983–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille-Reece, U.; Flynn, B.J.; Loré, K.; Koup, R.A.; Kedl, R.M.; Mattapallil, J.J.; Weiss, W.R.; Roederer, M.; Seder, R.A. HIV Gag protein conjugated to a Toll-like receptor 7/8 agonist improves the magnitude and quality of Th1 and CD8+ T cell responses in nonhuman primates. Proc. Natl. Acad. Sci. USA 2005, 102, 15190–15194. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
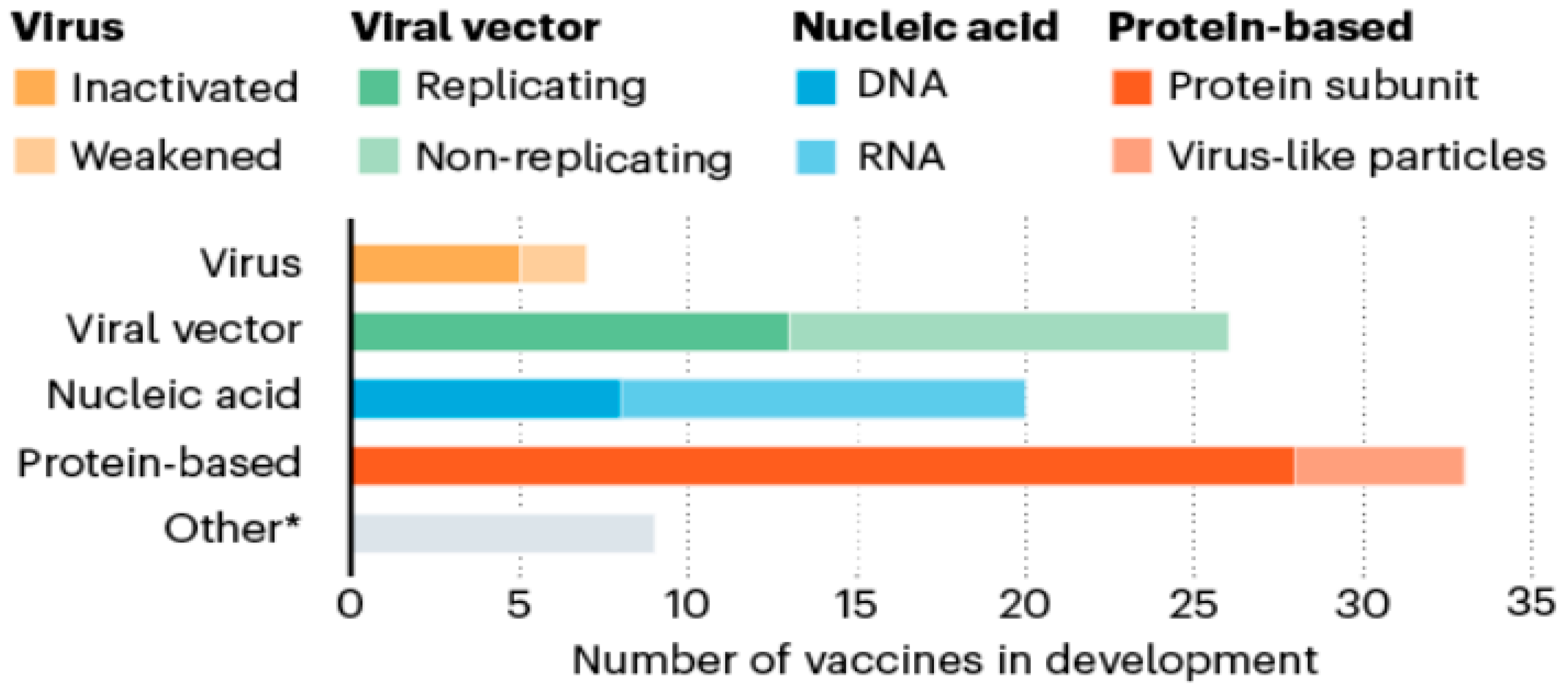{kind=link}
{kind=link}
| S. No | Position | Final B-cell Epitope | MHC-I | IC50 | MHC-II | IC50 |
|---|---|---|---|---|---|---|
| 1 | 404–424 | GDEVRQIAPGQTGKIADYNYK | VRQIAPGQT | 31.27 | VRQIAPGQT | 121 |
| 2 | 673–691 | SYQTQTNSPRRARSVASQS | QTQTNSPRR | 34.53 | QTQTNSPRR | 56 |
| 3 | 805–826 | ILPDPSKPSKRSFIEDLLFNKV | ILPDPSKPS | 23.04 | ILPDPSKPS | 157 |
| 4 | 14–36 | QCVNLTTRTQLPPAYTNSFTRGV | TQLPPAYTN | 11.95 | TQLPPAYTN | 121 |
| C1 adjuvant = HBHA adjuvant |
| EAAAKMAENPNIDDLPAPLLAALGAADLALATVNDLIANLRERAEETRAETRTRVEERRARLTKFQEDLPEQFIELRDKFTTEELRKAAEGYLEAATNRYNELVERGEAALQRLRSQTAFEDASARAEGYVDQAVELTQEALGTVASQTRAVGERAAKLVGIELEAAAKAKFVAAWTLKAAAGGGSGDEVRQIAPGQTGKIADYNYKGGGSSYQTQTNSPRRARSVASQSGGGSAKFVAAWTLKAAAGGGSILPDPSKPSKRSFIEDLLFNKVHEYGAEALERAGQCVNLTTRTQLPPAYTNSFTRGVHEYGAEALERAGAKFVAAWTLKAAAGGGS |
| C2 adjuvant = Beta defensin adjuvant |
| EAAAKGIINTLQKYYCRVRGGRCAVLSCLPKEEQIGKCSTRGRKCCRRKKEAAAKAKFVAAWTLKAAAGGGSGDEVRQIAPGQTGKIADYNYKGGGSILPDPSKPSKRSFIEDLLFNKVGGGSAKFVAAWTLKAAAGGGSSYQTQTNSPRRARSVASQSHEYGAEALERAGQCVNLTTRTQLPPAYTNSFTRGVHEYGAEALERAGAKFVAAWTLKAAAGGGS |
| C3 adjuvant = HBHA conserved |
| EAAAKMAENSNIDDIKAPLLAALGAADLALATVNELITNLRERAEETRRSRVEESRARLTKLQEDLPEQLTELREKFTAEELRKAAEGYLEAATSELVERGEAALERLRSQQSFEEVSARAEGYVDQAVELTQEALGTVASQVEGRAAKLVGIELEAAAKAKFVAAWTLKAAAGGGSSYQTQTNSPRRARSVASQSGGGSQCVNLTTRTQLPPAYTNSFTRGVGGGSAKFVAAWTLKAAAGGGSGDEVRQIAPGQTGKIADYNYKHEYGAEALERAGILPDPSKPSKRSFIEDLLFNKVHEYGAEALERAGAKFVAAWTLKAAAGGGS |
| C4 adjuvant = Ribosomal protein adjuvant |
| EAAAKMAKLSTDELLDAFKEMTLLELSDFVKKFEETFEVTAAAPVAVAAAGAAPAGAAVEAAEEQSEFDVILEAAGDKKIGVIKVVREIVSGLGLKEAKDLVDGAPKPLLEKVAKEAADEAKAKLEAAGATVTVKEAAAKAKFVAAWTLKAAAGGGSQCVNLTTRTQLPPAYTNSFTRGVGGGSGDEVRQIAPGQTGKIADYNYKGGGSAKFVAAWTLKAAAGGGSILPDPSKPSKRSFIEDLLFNKVHEYGAEALERAGSYQTQTNSPRRARSVASQSHEYGAEALERAGAKFVAAWTLKAAAGGGS |
| C5 adjuvant = flagellin adjuvant |
| EAAAKMAQVINTNSLSLLTQNNLNKSQSSLSSAIERLSSGLRINSAKDDAAGQAIANRFTSNIKGLTQASRNANDGISIAQTTEGALNEINNNLQRVRELSVQATNGTNSDSDLKSIQDEIQQRLEEIDRVSNQTQFNGVKVLSQDNQMKIQVGANDGETITIDLQKIDVKSLGLDGFNVEAAAKAKFVAAWTLKAAAGGGSGDEVRQIAPGQTGKIADYNYKGGGSSYQTQTNSPRRARSVASQSGGGSAKFVAAWTLKAAAGGGSILPDPSKPSKRSFIEDLLFNKVHEYGAEALERAGQCVNLTTRTQLPPAYTNSFTRGVHEYGAEALERAGAKFVAAWTLKAAAGGGS |
| S. No | Antigenicity (Threshold > 0.4) | Solubility | Allergenicity (Threshold −0.4) |
|---|---|---|---|
| C1 | 0.4987 | 0.837445 | −0.83923292 |
| C2 | 0.5230 | 0.887539 | −0.75524626 |
| C3 | 0.5147 | 0.858435 | −0.75971333 |
| C4 | 0.4687 | 0.852533 | 0.13431533 |
| C5 | 0.4846 | 0.520147 | 0.51140747 |
| S. No | Number of Amino Acids | Molecular Weight (Daltons) | Theoretical pI | Aliphatic Index | GRAVY | Instability Index |
|---|---|---|---|---|---|---|
| C1 | 337 | 35,906.03 | 6.00 | 76.44 | −0.431 | 39.46 (stable) |
| C2 | 223 | 23,438.58 | 9.88 | 64.13 | −0.439 | 36.96 (stable) |
| C3 | 328 | 34,787.80 | 5.61 | 79.39 | −0.399 | 44.66 (unstable) |
| C4 | 308 | 31,717.87 | 6.32 | 80.42 | −0.140 | 28.73 (stable) |
| C5 | 353 | 37,181.20 | 9.01 | 78.39 | −0.451 | 31.65 (stable) |
| S. No | Solution Number | Global Energy (Kcal/mol) | Attractive VdW | Repulsive VdW | ACE | HB |
|---|---|---|---|---|---|---|
| TLR4/C1 | 268 | −43.48 | −41.07 | 20.13 | 0.39 | −3.21 |
| TLR4/C2 | 250 | −48.43 | −37.87 | 14.87 | 1.01 | −3.09 |
| TLR4/C3 | 753 | −57.79 | −34.68 | 24.66 | −6.82 | −6.15 |
| TLR4/C4 | 58 | −49.06 | −32.34 | 3.16 | 6.95 | −1.79 |
| TLR4/C5 | 307 | −37.22 | −27.03 | 19.07 | −2.02 | −1.64 |
| TLR7/C1 | 94 | −65.88 | −37.52 | 18.56 | −7.02 | −3.37 |
| TLR7/C2 | 438 | −47.16 | −41.24 | 17.91 | 7.72 | −3.18 |
| TLR7/C3 | 256 | −43.91 | −26.80 | 6.60 | 2.39 | −1.62 |
| TLR7/C4 | 839 | −55.98 | −39.32 | 16.45 | −3.15 | −4.69 |
| TLR7/C5 | 620 | −47.34 | −29.54 | 18.96 | −3.22 | −1.92 |
| TLR8/C1 | 383 | −60.24 | −42.98 | 17.59 | −0.09 | −5.04 |
| TLR8/C2 | 273 | −57.44 | −42.20 | 21.45 | 2.39 | −6.75 |
| TLR8/C3 | 973 | −50.57 | −29.17 | 24.59 | −11.45 | −1.43 |
| TLR8/C4 | 280 | −39.20 | −34.23 | 10.77 | 5.27 | −3.89 |
| TLR8/C5 | 902 | −60.34 | −42.13 | 28.02 | 1.06 | −2.06 |
| HADDOCK score | −132.1 +/− 7.3 |
| Cluster size | 45 |
| RMSD from the overall lowest-energy structure | 0.5 +/− 0.3 |
| Van der Waals energy | −117.0 +/− 6.3 |
| Electrostatic energy | −331.2 +/− 49.7 |
| Desolvation energy | −75.8 +/− 6.4 |
| Restraints violation energy | 1269.5 +/− 72.06 |
| Buried Surface Area | 3552.9 +/− 125.7 |
| Z-Score | −2.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, N.; Ali, F.; Basharat, Z.; Shehroz, M.; Khan, M.K.; Jeandet, P.; Nepovimova, E.; Kuca, K.; Khan, H. Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines 2020, 8, 423. https://doi.org/10.3390/vaccines8030423
Rahman N, Ali F, Basharat Z, Shehroz M, Khan MK, Jeandet P, Nepovimova E, Kuca K, Khan H. Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines. 2020; 8(3):423. https://doi.org/10.3390/vaccines8030423
Chicago/Turabian StyleRahman, Noor, Fawad Ali, Zarrin Basharat, Muhammad Shehroz, Muhammad Kazim Khan, Philippe Jeandet, Eugenie Nepovimova, Kamil Kuca, and Haroon Khan. 2020. "Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach" Vaccines 8, no. 3: 423. https://doi.org/10.3390/vaccines8030423
APA StyleRahman, N., Ali, F., Basharat, Z., Shehroz, M., Khan, M. K., Jeandet, P., Nepovimova, E., Kuca, K., & Khan, H. (2020). Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines, 8(3), 423. https://doi.org/10.3390/vaccines8030423