Hepatitis E Virus Mediates Renal Injury via the Interaction between the Immune Cells and Renal Epithelium

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Subjects
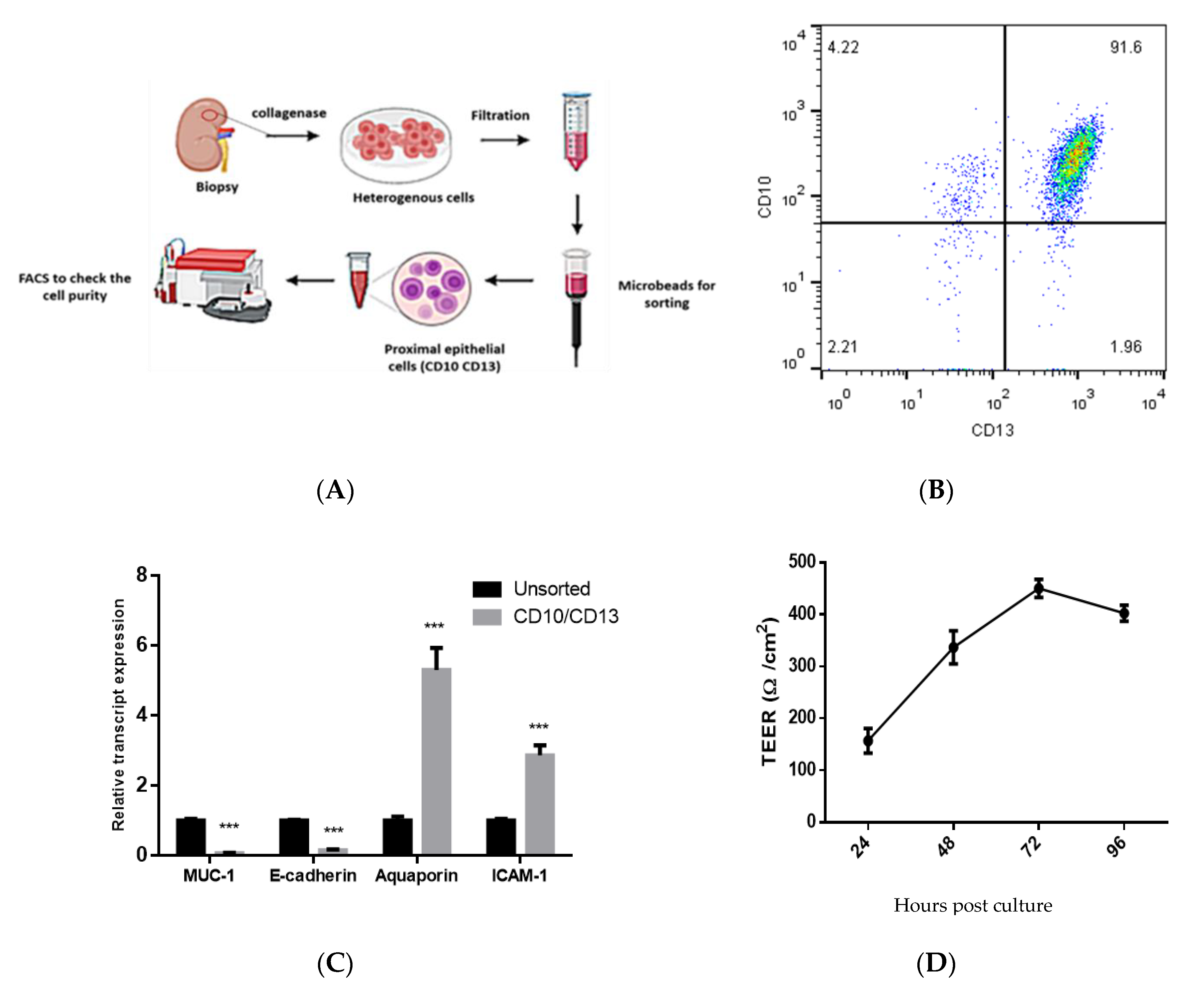
2.2. Isolation and Characterization of Double-Positive Proximal Tubular (PT) Epithelial Cells
2.3. Measurements of Trans-Epithelial Electrical Resistance (TEER)
2.4. Infection of CD10+/CD13+ PT Epithelial Cells with HEV Preparation
2.5. Assessment of HEV Viral Load by qPCR
2.6. Detection of Intracellular and Extracellular HEV Capsid Protein
2.7. CoCulture of CD10+/CD13+ PT Epithelial Cells with PBMCs
2.8. Test the Effect of HEV Infection on the Transcriptome of CD10/CD13 PT Epithelial Cells
2.9. LDH Assay
2.10. Measurement the Level of Inflammatory Cytokines Released after HEV Infection
2.11. Treatment of PT Cells with IFN-γ
2.12. Statistics
3. Results
3.1. Isolation and Characterization of Primary Human PT Epithelial Cells
3.2. Infection of the CD10+/CD13+ PT Epithelial Cells with HEV Inoculum
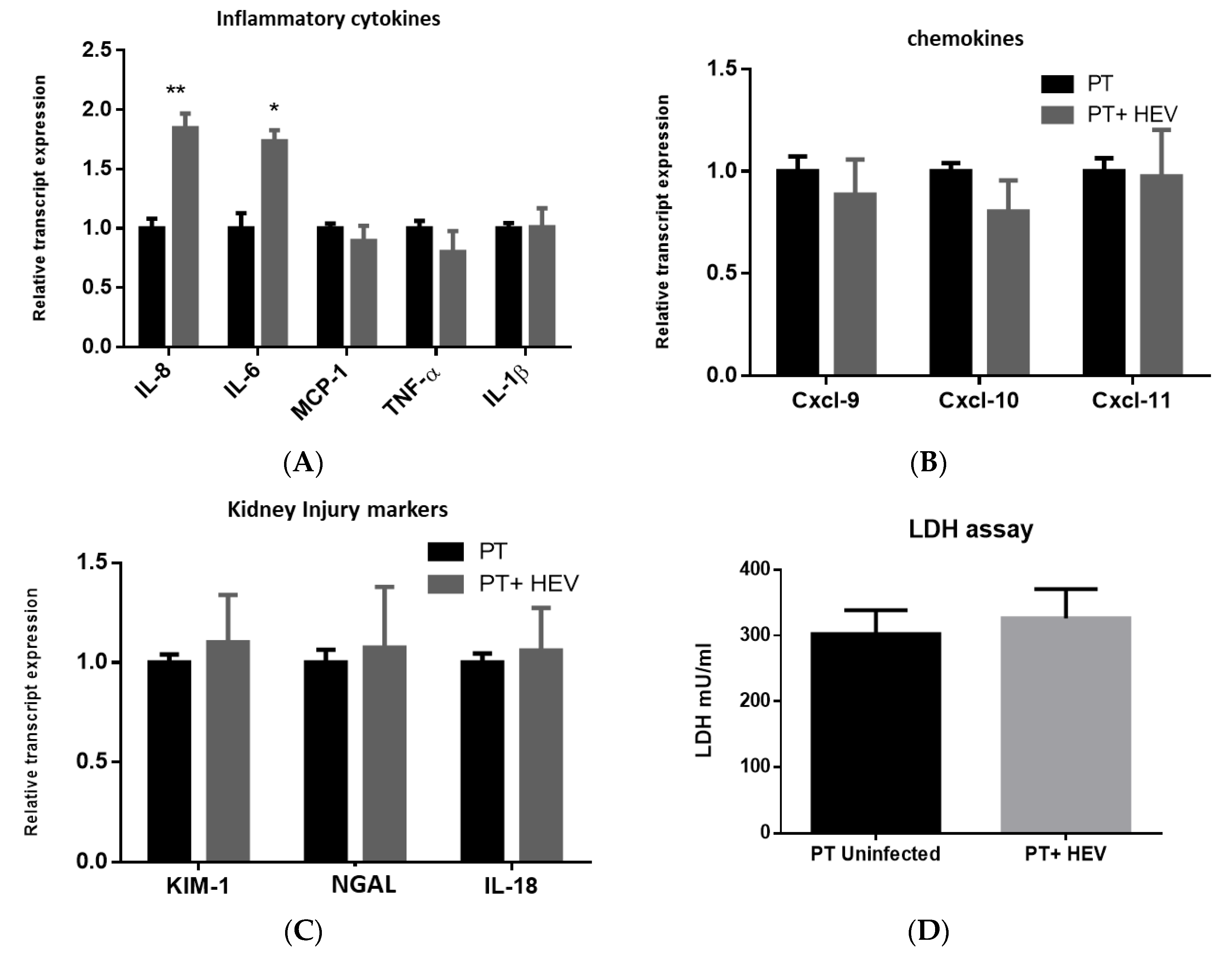
3.3. HEV Infection Alone Slightly Elevated the Inflammatory Response with No Effect on the Transcription of Chemokines nor Kidney Injury Markers
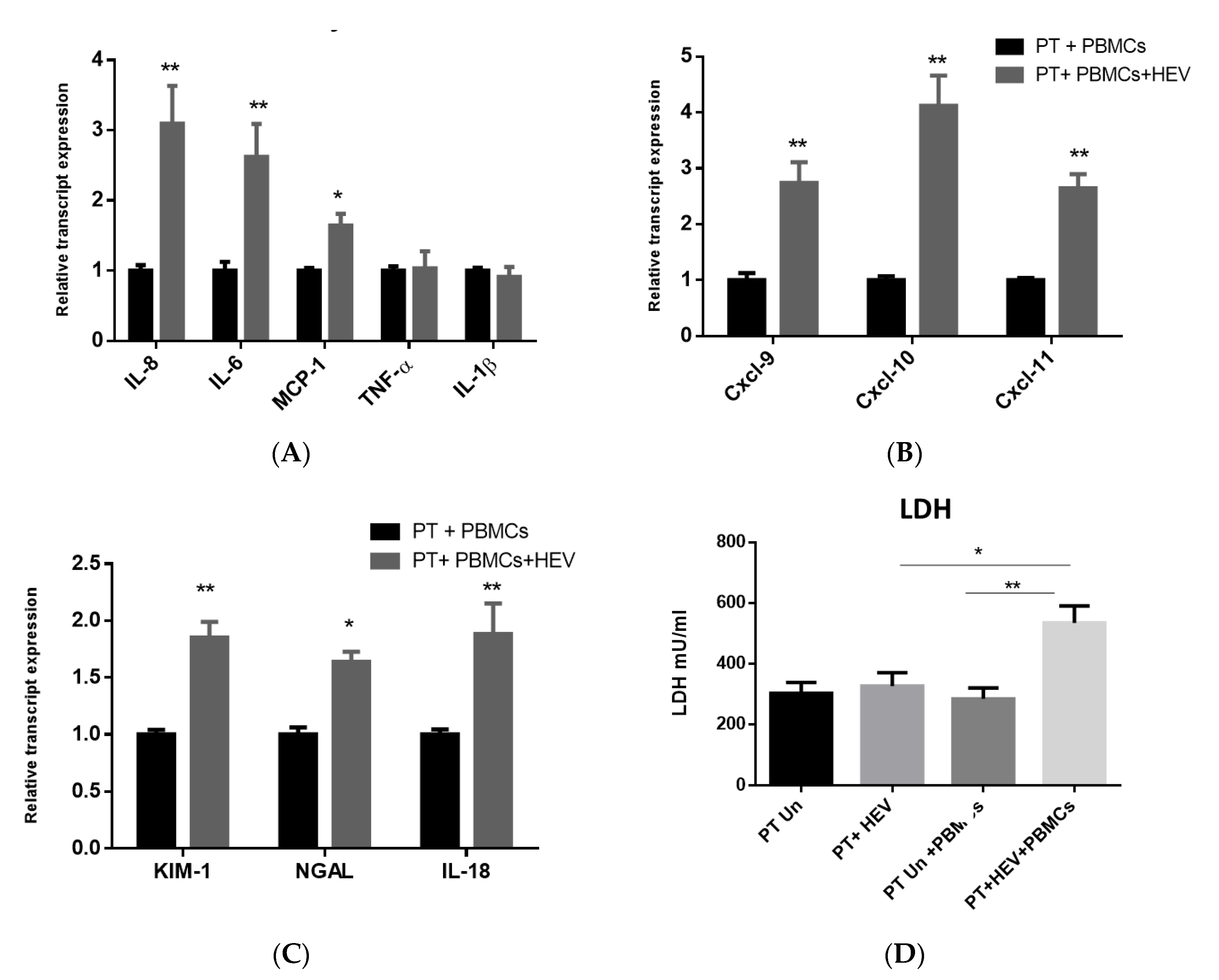
3.4. The CoCulture of PBMCs with the HEV-Infected PT Epithelial Cells Increased the Expression of Inflammatory Cytokines, Chemokines, and Kidney Injury Markers Produced from the Epithelium
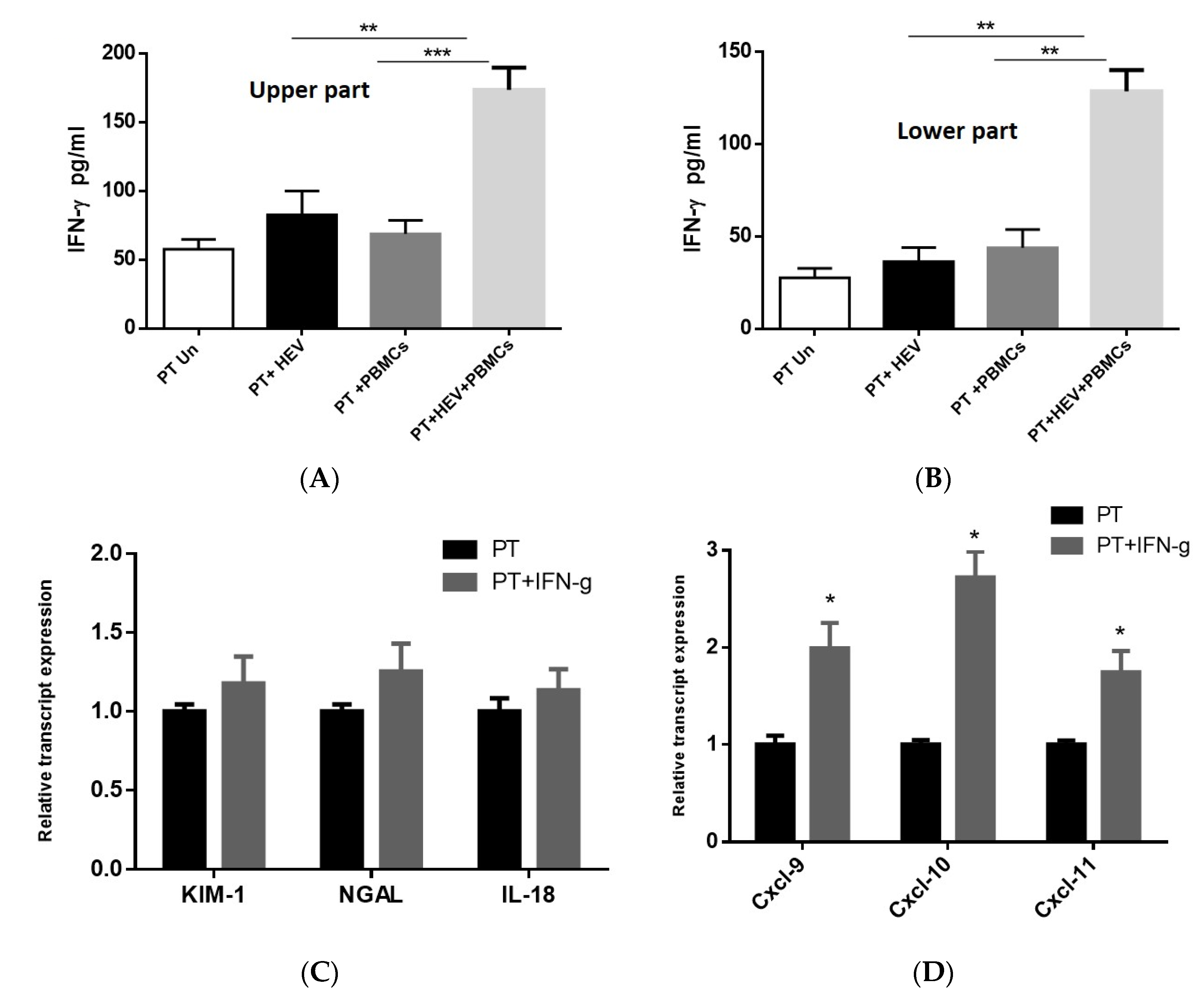
3.5. Exacerbation of the Inflammatory Response Was Dependable on IFN-γ Produced from the PBMCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Vercouter, A.S.; Abdelwahab, S.F.; Vercauteren, K.; Meuleman, P. Is hepatitis E virus an emerging problem in industrialized countries? Hepatology 2015, 62, 1883–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, I.M.; Elkhawaga, A.A.; El-Mokhtar, M.A. In vivo models for studying Hepatitis E virus infection; Updates and applications. Virus Res. 2019, 274, 197765. [Google Scholar] [CrossRef]
- Sayed, I.M.; Vercauteren, K.; Abdelwahab, S.F.; Meuleman, P. The emergence of hepatitis E virus in Europe. Future Virol. 2015, 10, 763–778. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.J.; Purcell, R.H.; Halbur, P.G.; Lehman, J.R.; Webb, D.M.; Tsareva, T.S.; Haynes, J.S.; Thacker, B.J.; Emerson, S.U. A novel virus in swine is closely related to the human hepatitis E virus. Proc. Natl. Acad. Sci. USA 1997, 94, 9860–9865. [Google Scholar] [CrossRef] [Green Version]
- Pavio, N.; Meng, X.J.; Doceul, V. Zoonotic origin of hepatitis E. Curr. Opin. Virol. 2015, 10, 34–41. [Google Scholar] [CrossRef]
- Kenney, S.P. The Current Host Range of Hepatitis E Viruses. Viruses 2019, 11, 452. [Google Scholar] [CrossRef] [Green Version]
- Domanović, D.; Tedder, R.; Blümel, J.; Zaaijer, H.; Gallian, P.; Niederhauser, C.; Sauleda Oliveras, S.; O’Riordan, J.; Boland, F.; Harritshøj, L.; et al. Hepatitis E and blood donation safety in selected European countries: A shift to screening? Eurosurveillance 2017, 22. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kumar, A.; Kar, P.; Agarwal, S.; Ramji, S.; Husain, S.A.; Prasad, S.; Sharma, S. Risk factors for vertical transmission of hepatitis E virus infection. J. Viral Hepat. 2017, 24, 1067–1075. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef]
- Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Tedder, R.S.; Ijaz, S. Persistent carriage of hepatitis E virus in patients with HIV infection. N. Eng. J. Med. 2009, 361, 1025–1027. [Google Scholar] [CrossRef]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Péron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Eng. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [Green Version]
- Pischke, S.; Hartl, J.; Pas, S.D.; Lohse, A.W.; Jacobs, B.C.; Van der Eijk, A.A. Hepatitis E virus: Infection beyond the liver? J. Hepatol. 2017, 66, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Mansuy, J.M.; Esposito, L.; Legrand-Abravanel, F.; Peron, J.M.; Durand, D.; Rostaing, L.; Izopet, J. Acute hepatitis and renal function impairment related to infection by hepatitis E virus in a renal allograft recipient. Am. J. Kidney Dis. 2005, 45, 193–196. [Google Scholar] [CrossRef]
- Kamar, N.; Weclawiak, H.; Guilbeau-Frugier, C.; Legrand-Abravanel, F.; Cointault, O.; Ribes, D.; Esposito, L.; Cardeau-Desangles, I.; Guitard, J.; Sallusto, F.; et al. Hepatitis E Virus and the Kidney in Solid-Organ Transplant Patients. Transplantation 2012, 93, 617–623. [Google Scholar] [CrossRef]
- Nayak, S.; Sharma, M.; Kataria, A.; Tiwari, S.C.; Rastogi, A.; Mukund, A. Cholemic Nephrosis from Acute Hepatitis E Virus Infection: A Forgotten Entity? Indian J. Nephrol. 2018, 28, 250–251. [Google Scholar] [CrossRef]
- Wang, L.; Xia, J.; Wang, L.; Wang, Y. Experimental infection of rabbits with genotype 3 hepatitis E virus produced both chronicity and kidney injury. Gut 2017, 66, 561–562. [Google Scholar] [CrossRef]
- Huang, F.; Yang, C.; Zhou, X.; Yu, W.; Pan, Q. Rhesus macaques persistently infected with hepatitis E shed virus into urine. J. Hepatol. 2016, 64, 1446–1447. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Zhao, C.; Huang, W.; Harrison, T.J.; Zhang, H.; Geng, K.; Wang, Y. Detection and assessment of infectivity of hepatitis E virus in urine. J. Hepatol. 2016, 64, 37–43. [Google Scholar] [CrossRef]
- Marion, O.; Capelli, N.; Lhomme, S.; Dubois, M.; Pucelle, M.; Abravanel, F.; Kamar, N.; Izopet, J. Hepatitis E virus genotype 3 and capsid protein in the blood and urine of immunocompromised patients. J. Infect. 2019, 78, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Ozkok, A.; Yildiz, A. Hepatitis C virus associated glomerulopathies. World J. Gastroenterol. 2014, 20, 7544–7554. [Google Scholar] [CrossRef] [PubMed]
- Salter, T.; Burton, H.; Douthwaite, S.; Newsholme, W.; Horsfield, C.; Hilton, R. Immune Complex Mediated Glomerulonephritis with Acute Thrombotic Microangiopathy following Newly Detected Hepatitis B Virus Infection in a Kidney Transplant Recipient. Case Rep. Transplant. 2016, 2016, 3152495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, M.A.; Sarwar, S.; Chaudry, M.A.; Maqbool, B.; Khalil, Z.; Tan, J.; Yaqub, S.; Hussain, S.A. Acute kidney injury in dengue virus infection. Clin. Kidney J. 2012, 5, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Tang, L.; Tang, H.; Pu, J.; Gong, S.; Fang, D.; Zhang, H.; Li, Y.P.; Zhu, X.; Wang, W.; et al. Zika Virus Infection Induces Acute Kidney Injury Through Activating NLRP3 Inflammasome Via Suppressing Bcl-2. Front. Immunol. 2019, 10, 1925. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, M.S.; Kashani, K.B. Biomarkers for Early Detection of Acute Kidney Injury. J. Appl. Lab. Med. 2019, 2, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Pischke, S.; Behrendt, P.; Manns, M.P.; Wedemeyer, H. HEV-associated cryoglobulinaemia and extrahepatic manifestations of hepatitis E. Lancet Infect. Dis. 2014, 14, 678–679. [Google Scholar] [CrossRef]
- Karki, P.; Malik, S.; Mallick, B.; Sharma, V.; Rana, S.S. Massive Hemolysis Causing Renal Failure in Acute Hepatitis E Infection. J. Clin. Transl. Hepatol. 2016, 4, 345–347. [Google Scholar] [CrossRef] [Green Version]
- El-Mokhtar, M.A.; Othman, E.R.; Khashbah, M.Y.; Ismael, A.; Ghaliony, M.A.; Seddik, M.I.; Sayed, I.M. Evidence of the Extrahepatic Replication of Hepatitis E Virus in Human Endometrial Stromal Cells. Pathogens 2020, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.E.; El-Badawy, O.; Afifi, N.A.; Eldin, A.S.; Hassan, E.A.; Halby, H.M.; El-Mokhtar, M.A. Role of T-Helper 9 Cells in Chronic Hepatitis C-Infected Patients. Viruses 2018, 10, 341. [Google Scholar] [CrossRef] [Green Version]
- El-Mokhtar, M.A.; Elgendy, S.G.; Eldin, A.S. Hepatitis C Virus Affects Tuberculosis-Specific T Cells in HIV-Negative Patients. Viruses 2020, 12, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Hauwaert, C.; Savary, G.; Gnemmi, V.; Glowacki, F.; Pottier, N.; Bouillez, A.; Maboudou, P.; Zini, L.; Leroy, X.; Cauffiez, C.; et al. Isolation and characterization of a primary proximal tubular epithelial cell model from human kidney by CD10/CD13 double labeling. PLoS ONE 2013, 8, e66750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, I.M.; Seddik, M.I. Replication of Hepatitis E Virus (HEV) in Primary Human-Derived Monocytes and Macrophages In Vitro. Vaccines 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Foquet, L.; Verhoye, L.; Abravanel, F.; Farhoudi, A.; Leroux-Roels, G.; Izopet, J.; Meuleman, P. Transmission of hepatitis E virus infection to human-liver chimeric FRG mice using patient plasma. Antivir. Res. 2017, 141, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920–929. [Google Scholar] [CrossRef]
- Sayed, I.M.; Meuleman, P. Murine Tissues of Human Liver Chimeric Mice Are Not Susceptible to Hepatitis E Virus Genotypes 1 and 3. J. Infect. Dis. 2017, 216, 919–920. [Google Scholar] [CrossRef]
- Sayed, I.M.; Elkhawaga, A.A.; El-Mokhtar, M.A. Circulation of hepatitis E virus (HEV) and/or HEV-like agent in non-mixed dairy farms could represent a potential source of infection for Egyptian people. Int. J. Food Microbiol. 2020, 317, 108479. [Google Scholar] [CrossRef]
- Sayed, I.M.; Suarez, K.; Lim, E.; Singh, S.; Pereira, M.; Ibeawuchi, S.R.; Katkar, G.; Dunkel, Y.; Mittal, Y.; Chattopadhyay, R.; et al. Host engulfment pathway controls inflammation in inflammatory bowel disease. FEBS J. 2020. [Google Scholar] [CrossRef]
- Lin, Q.; Song, Y.; Zhu, X.; Yang, S.; Zheng, J. Expressions of CXCL9, CXCL10 and CXCL11 in renal tubular epithelial cells induced by IFN-γ. Chin. J. Cell. Mol. Immunol. 2013, 29, 137–140. [Google Scholar]
- Arai, Y.; Takahashi, D.; Asano, K.; Tanaka, M.; Oda, M.; Ko, S.B.H.; Ko, M.S.H.; Mandai, S.; Nomura, N.; Rai, T.; et al. Salt suppresses IFNγ inducible chemokines through the IFNγ-JAK1-STAT1 signaling pathway in proximal tubular cells. Sci. Rep. 2017, 7, 46580. [Google Scholar] [CrossRef] [Green Version]
- Meda Spaccamela, V.; Valencia, R.G.; Pastukhov, O.; Duppenthaler, A.; Dettmer, M.S.; Erb, J.; Steiner, U.C.; Hillinger, S.; Speckmann, C.; Ehl, S.; et al. High Levels of IL-18 and IFN-γ in Chronically Inflamed Tissue in Chronic Granulomatous Disease. Front. Immunol. 2019, 10, 2236. [Google Scholar] [CrossRef] [PubMed]
- Guinault, D.; Ribes, D.; Delas, A.; Milongo, D.; Abravanel, F.; Puissant-Lubrano, B.; Izopet, J.; Kamar, N. Hepatitis E Virus-Induced Cryoglobulinemic Glomerulonephritis in a Nonimmunocompromised Person. Am. J. Kidney Dis. 2016, 67, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Ali, G.; Kumar, M.; Bali, S.; Wadhwa, W. Hepatitis E associated immune thrombocytopaenia and membranous glomerulonephritis. Indian J. Nephrol. 2001, 11, 70. [Google Scholar]
- Wang, Y.; Chen, G.; Pan, Q.; Zhao, J. Chronic Hepatitis E in a Renal Transplant Recipient: The First Report of Genotype 4 Hepatitis E Virus Caused Chronic Infection in Organ Recipient. Gastroenterology 2018, 154, 1199–1201. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, G. Renal involvement in hepatitis C infection: Cryoglobulinemic glomerulonephritis. Kidney Int. 1998, 54, 650–671. [Google Scholar] [CrossRef] [Green Version]
- Verschuuren, E.; Haagsma, E.; Zijlstra, J.; Stegeman, C.A. Non-oliguric acute renal failure associated with hepatitis E. Nephrol. Dial. Transplant. 1997, 12, 799–801. [Google Scholar] [CrossRef] [Green Version]
- Baer, P.C.; Bereiter-Hahn, J.; Schubert, R.; Geiger, H. Differentiation status of human renal proximal and distal tubular epithelial cells in vitro: Differential expression of characteristic markers. Cells Tissues Organs 2006, 184, 16–22. [Google Scholar] [CrossRef]
- Williams, T.P.; Kasorndorkbua, C.; Halbur, P.G.; Haqshenas, G.; Guenette, D.K.; Toth, T.E.; Meng, X.J. Evidence of extrahepatic sites of replication of the hepatitis E virus in a swine model. J. Clin. Microbiol. 2001, 39, 3040–3046. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.T.; Shukla, P.; Torian, U.; Faulk, K.; Emerson, S.U. Hepatitis E virus genotype 1 infection of swine kidney cells in vitro is inhibited at multiple levels. J. Virol. 2014, 88, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Sayed, I.M.; Meuleman, P. Updates in Hepatitis E virus (HEV) field; lessons learned from human liver chimeric mice. Rev. Med. Virol. 2020, 30, e2086. [Google Scholar] [CrossRef]
- Sayed, I.M. Hepatic immune response against Hepatitis E virus genotype 1 infection among animal models. J. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Vercouter, A.S.; Meuleman, P. Hepatitis E virus in acute liver failure: An unusual suspect? Hepatology 2016, 64, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Hino, S.; Ri, J.; Sakai, K.; Nagare, Y.; Kawanishi, M.; Niki, K.; Funauchi, M.; Matsumura, I. Lipopolysaccharide-Induced Acute Kidney Injury Is Dependent on an IL-18 Receptor Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozaki, Y.; Kinoshita, K.; Yano, T.; Asato, K.; Shiga, T.; Hino, S.; Niki, K.; Nagare, Y.; Kishimoto, K.; Shimazu, H.; et al. Signaling through the interleukin-18 receptor α attenuates inflammation in cisplatin-induced acute kidney injury. Kidney Int. 2012, 82, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Saravanabalaji, S.; Tripathy, A.S.; Dhoot, R.R.; Chadha, M.S.; Kakrani, A.L.; Arankalle, V.A. Viral load, antibody titers and recombinant open reading frame 2 protein-induced TH1/TH2 cytokines and cellular immune responses in self-limiting and fulminant hepatitis e. Intervirology 2009, 52, 78–85. [Google Scholar] [CrossRef]
- Kumar, A.; Devi, S.G.; Kar, P.; Agarwal, S.; Husain, S.A.; Gupta, R.K.; Sharma, S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014, 65, 95–104. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Mokhtar, M.A.; Seddik, M.I.; Osman, A.; Adel, S.; Abdel Aziz, E.M.; Mandour, S.A.; Mohammed, N.; Zarzour, M.A.; Abdel-Wahid, L.; Radwan, E.; et al. Hepatitis E Virus Mediates Renal Injury via the Interaction between the Immune Cells and Renal Epithelium. Vaccines 2020, 8, 454. https://doi.org/10.3390/vaccines8030454
El-Mokhtar MA, Seddik MI, Osman A, Adel S, Abdel Aziz EM, Mandour SA, Mohammed N, Zarzour MA, Abdel-Wahid L, Radwan E, et al. Hepatitis E Virus Mediates Renal Injury via the Interaction between the Immune Cells and Renal Epithelium. Vaccines. 2020; 8(3):454. https://doi.org/10.3390/vaccines8030454
Chicago/Turabian StyleEl-Mokhtar, Mohamed A., Mohamed Ismail Seddik, Asmaa Osman, Sara Adel, Essam M. Abdel Aziz, Sahar A. Mandour, Nasreldin Mohammed, Mohamed A. Zarzour, Lobna Abdel-Wahid, Eman Radwan, and et al. 2020. "Hepatitis E Virus Mediates Renal Injury via the Interaction between the Immune Cells and Renal Epithelium" Vaccines 8, no. 3: 454. https://doi.org/10.3390/vaccines8030454
APA StyleEl-Mokhtar, M. A., Seddik, M. I., Osman, A., Adel, S., Abdel Aziz, E. M., Mandour, S. A., Mohammed, N., Zarzour, M. A., Abdel-Wahid, L., Radwan, E., & Sayed, I. M. (2020). Hepatitis E Virus Mediates Renal Injury via the Interaction between the Immune Cells and Renal Epithelium. Vaccines, 8(3), 454. https://doi.org/10.3390/vaccines8030454