Control of Cytoskeletal Dynamics by β-Arrestin1/Myosin Vb Signaling Regulates Endosomal Sorting and Scavenging Activity of the Atypical Chemokine Receptor ACKR2

 ,
,  ,
,  , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Antibodies
2.2. Plasmids
2.3. Cell Culture and Transfection
2.4. Chemokine Scavenging and ACKR2 Membrane Expression Analysis
2.5. Immunofluorescence and Confocal Microscopy Analysis
2.6. Apoptosis Analysis
2.7. Statistical Analysis
3. Results
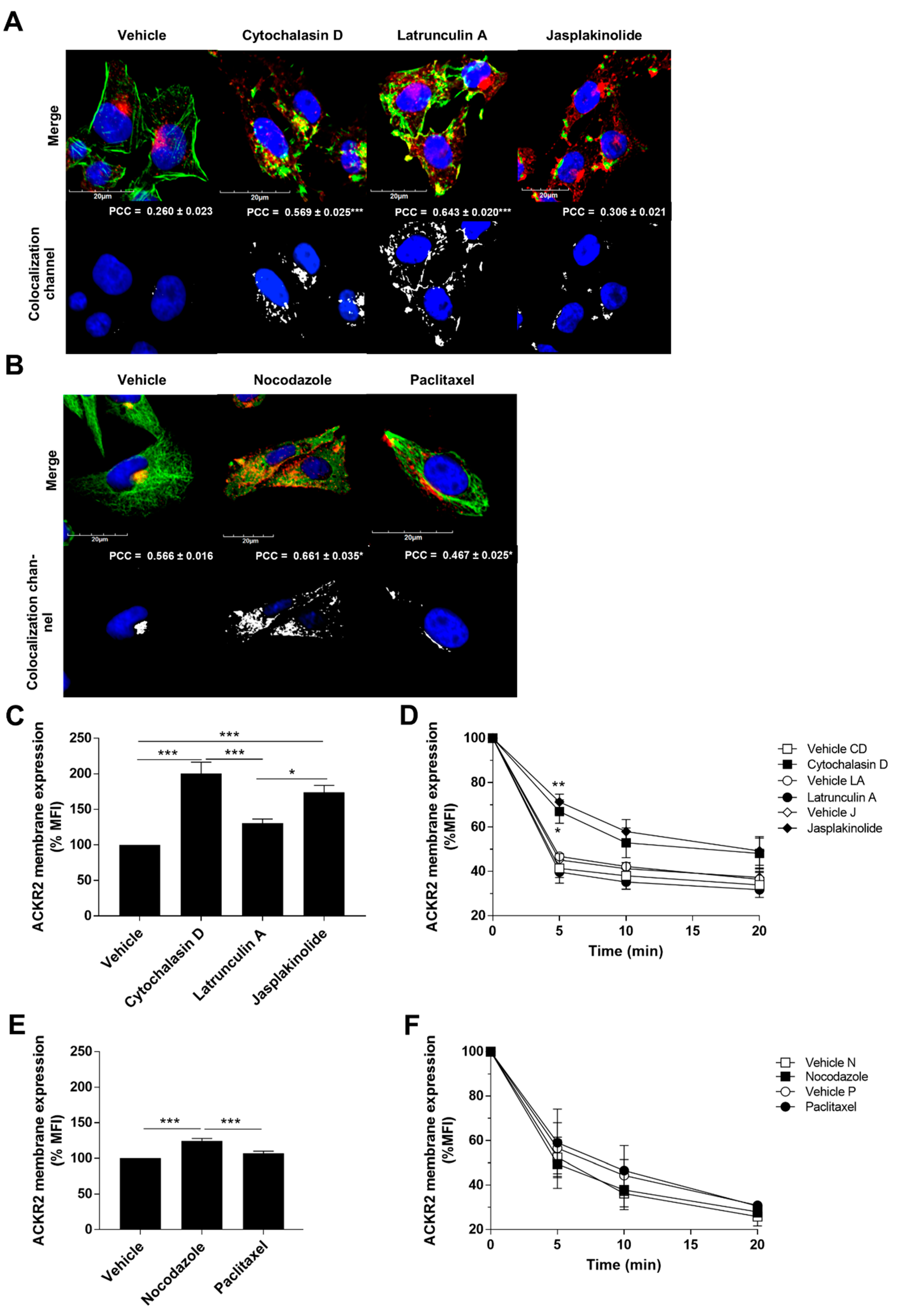
3.1. ACKR2 Constitutive Recycling Is Regulated by Cytoskeletal Dynamics
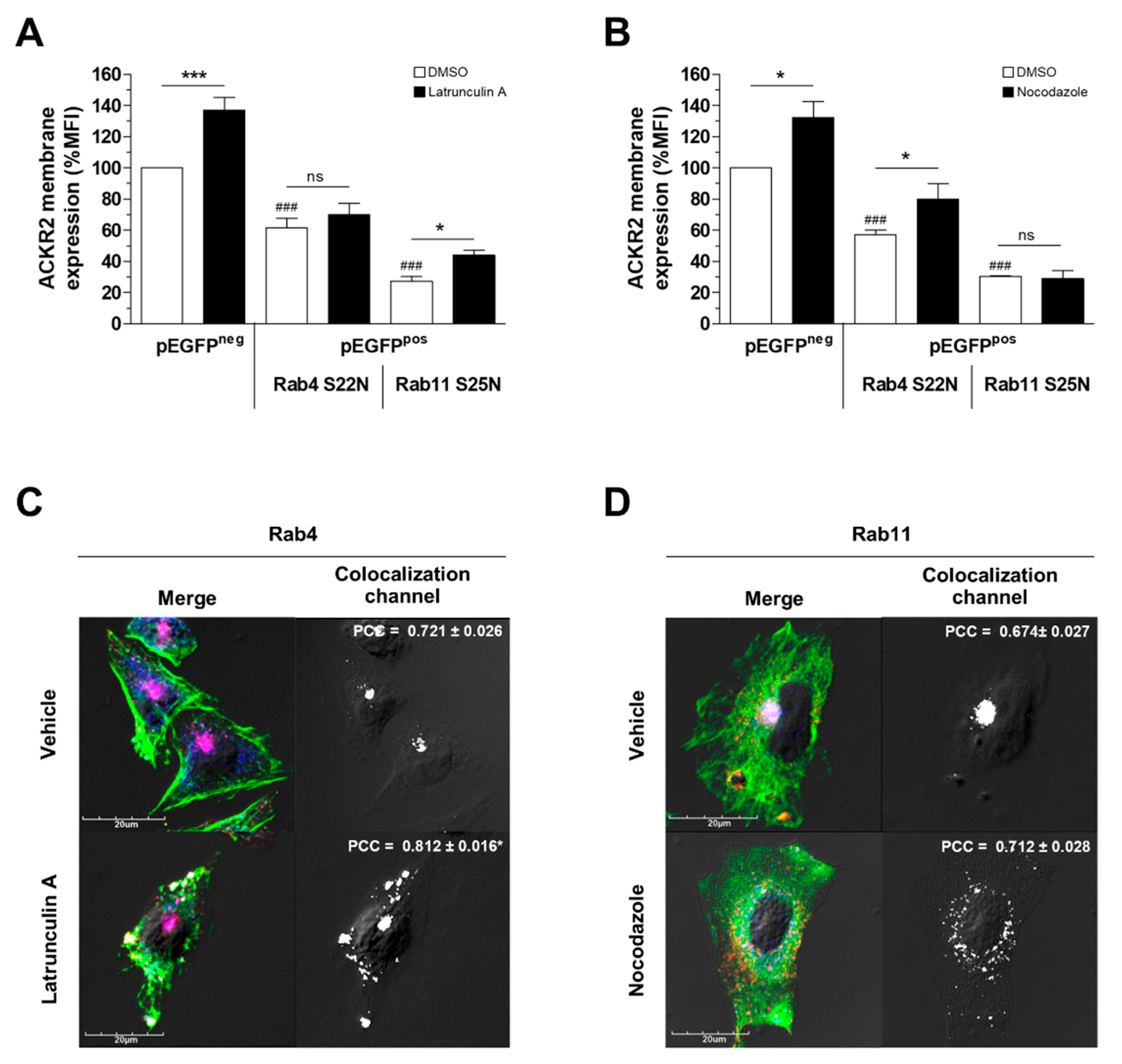
3.2. Alteration of Cytoskeletal Dynamics Causes ACKR2 Missorting into Recycling Compartments
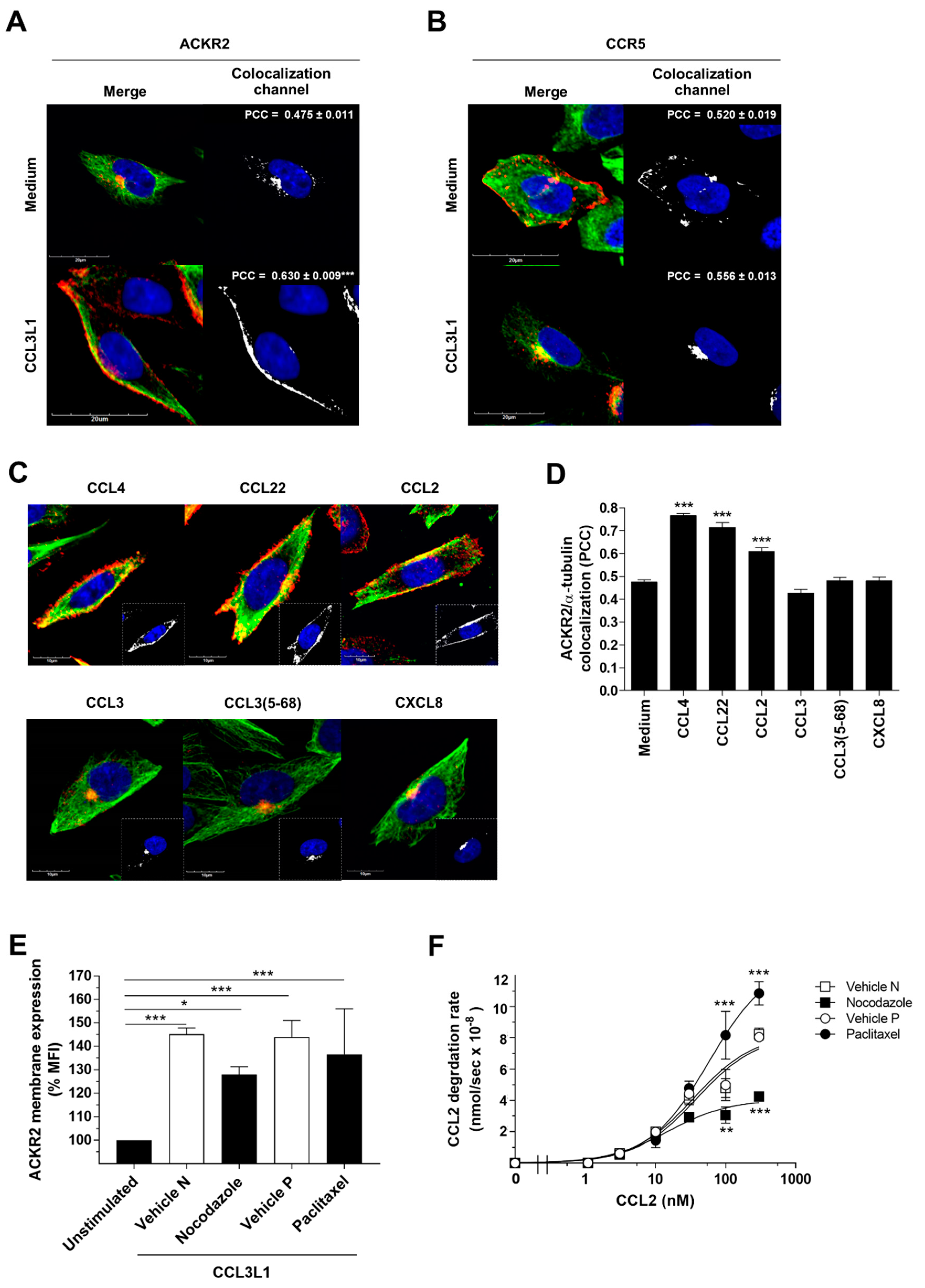
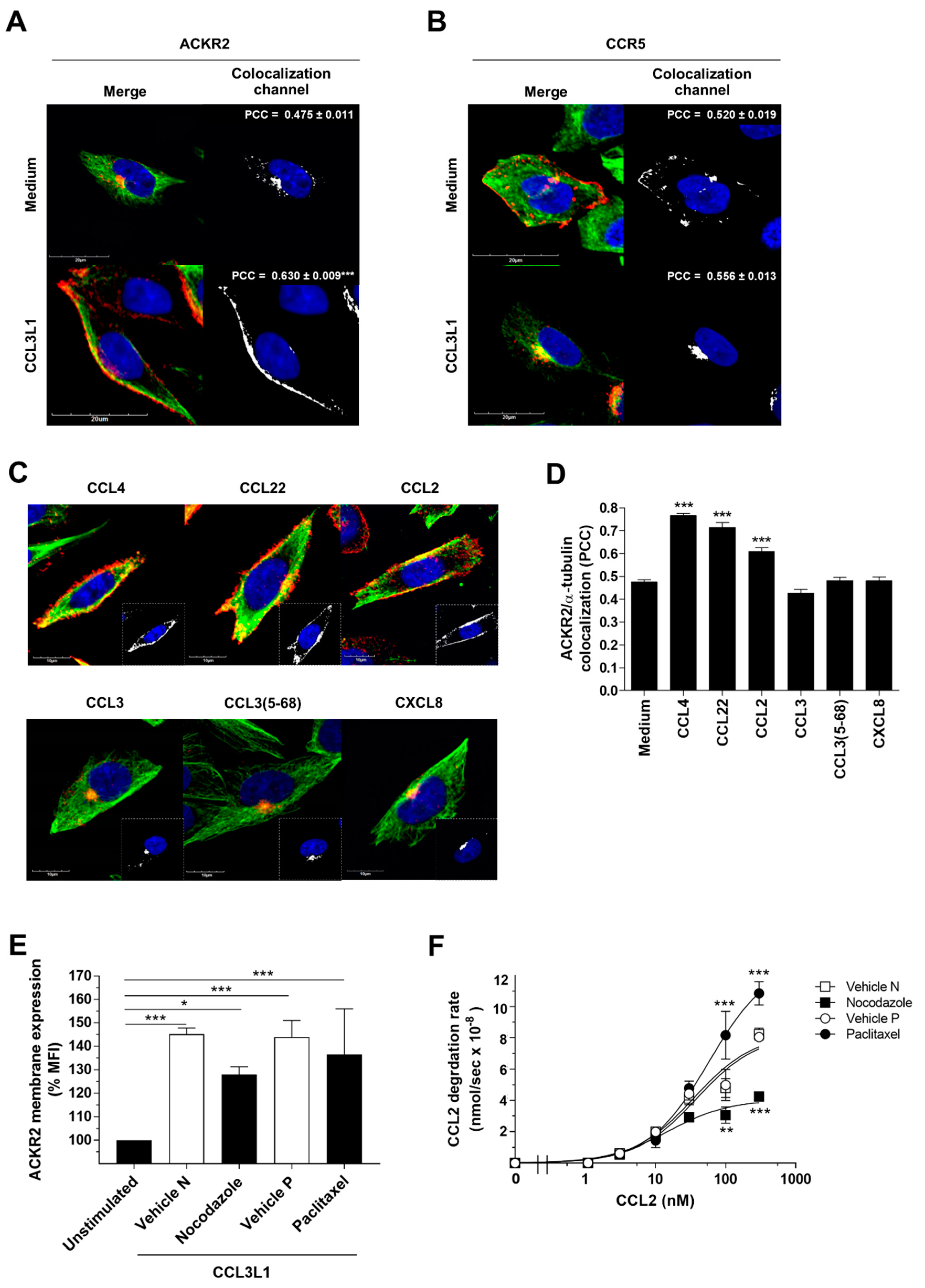
3.3. The Scavenger Function of ACKR2 Requires Ligand-Induced Rearrangement of Microtubules
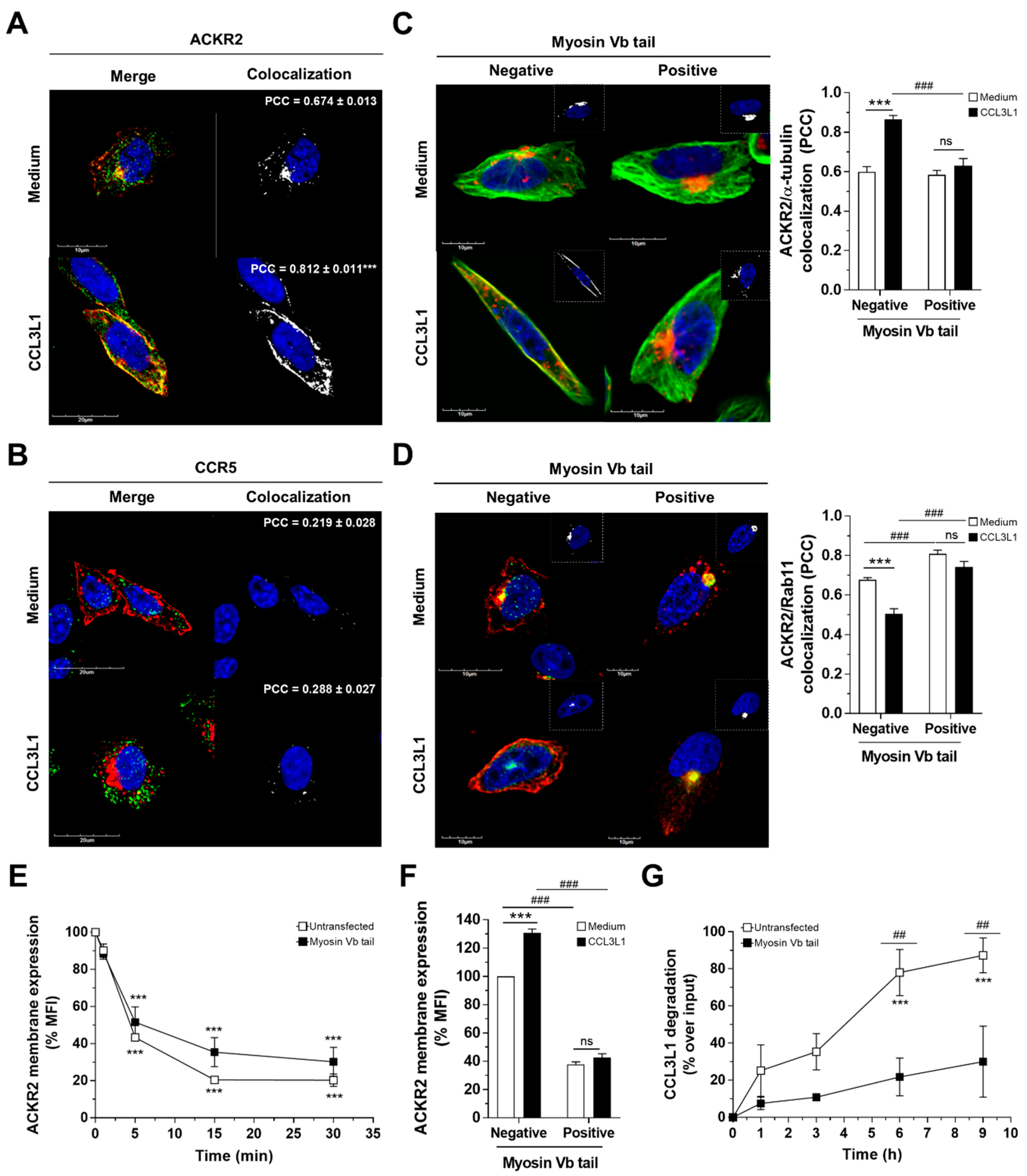
3.4. Myosin Vb Sustains ACKR2 Upregulation and Chemokine Degradation
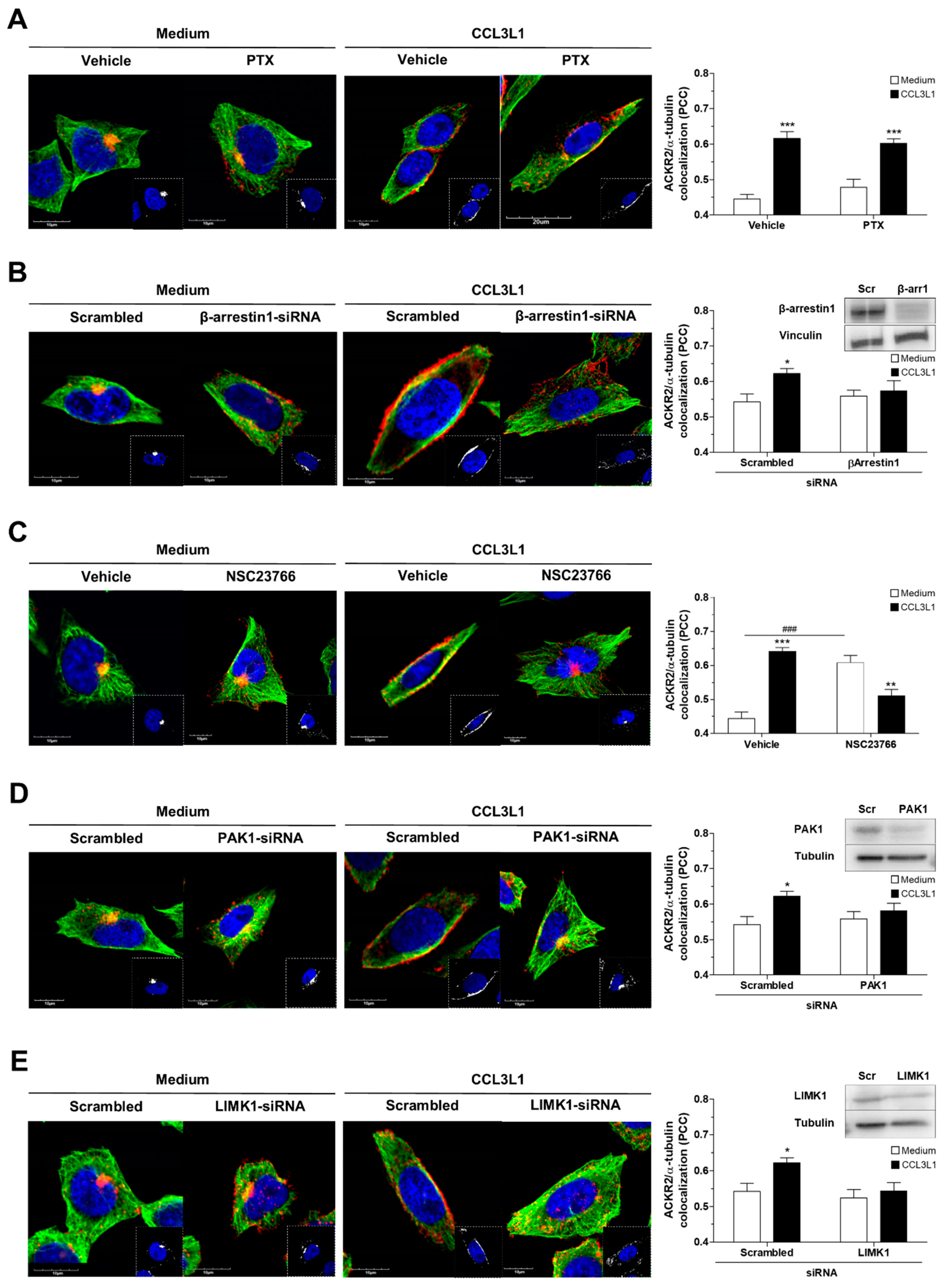
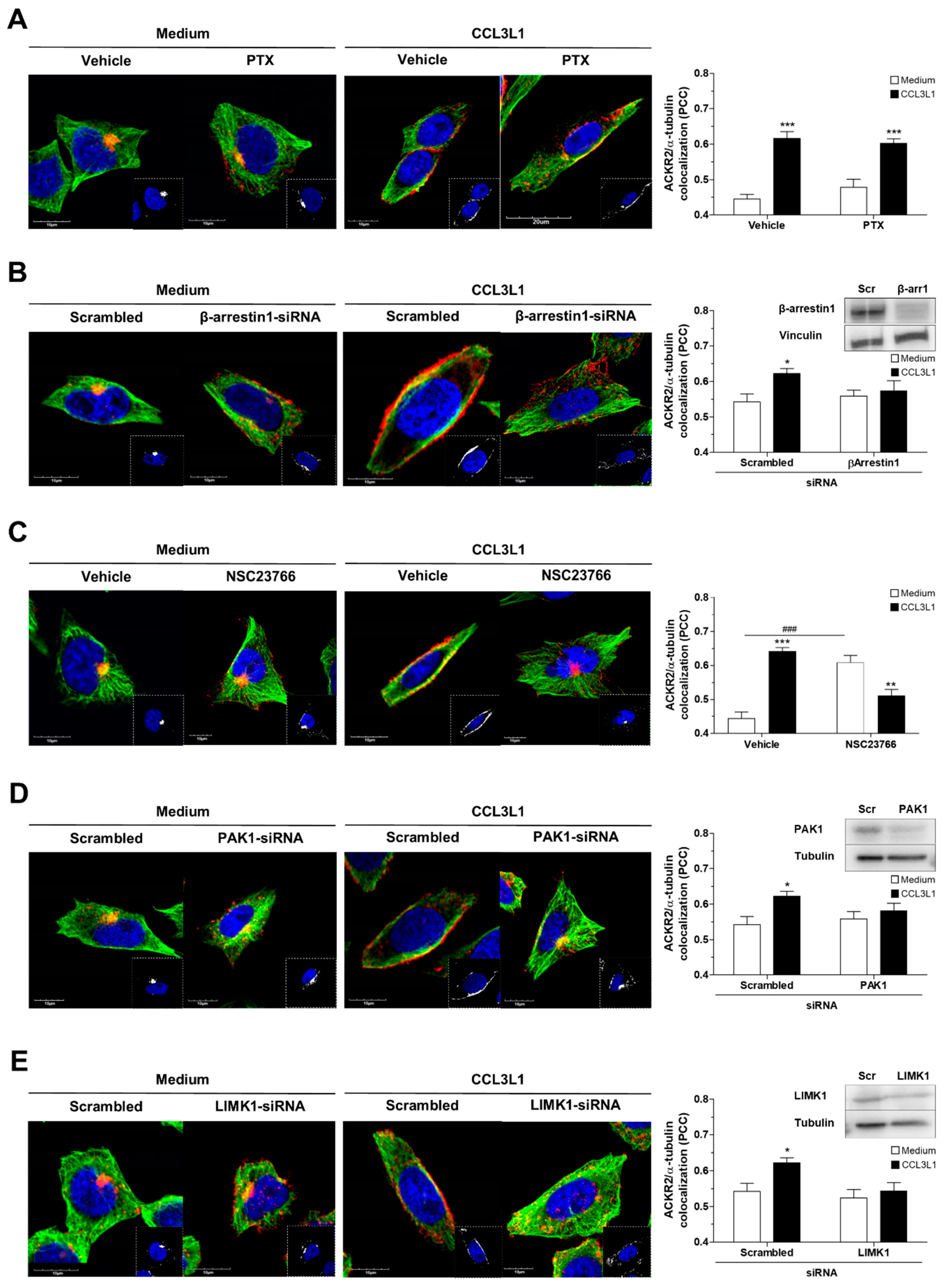
3.5. ACKR2 Promotes Microtubule Rearrangement through a G Protein-Independent β-Arrestin1-Dependent Pathway
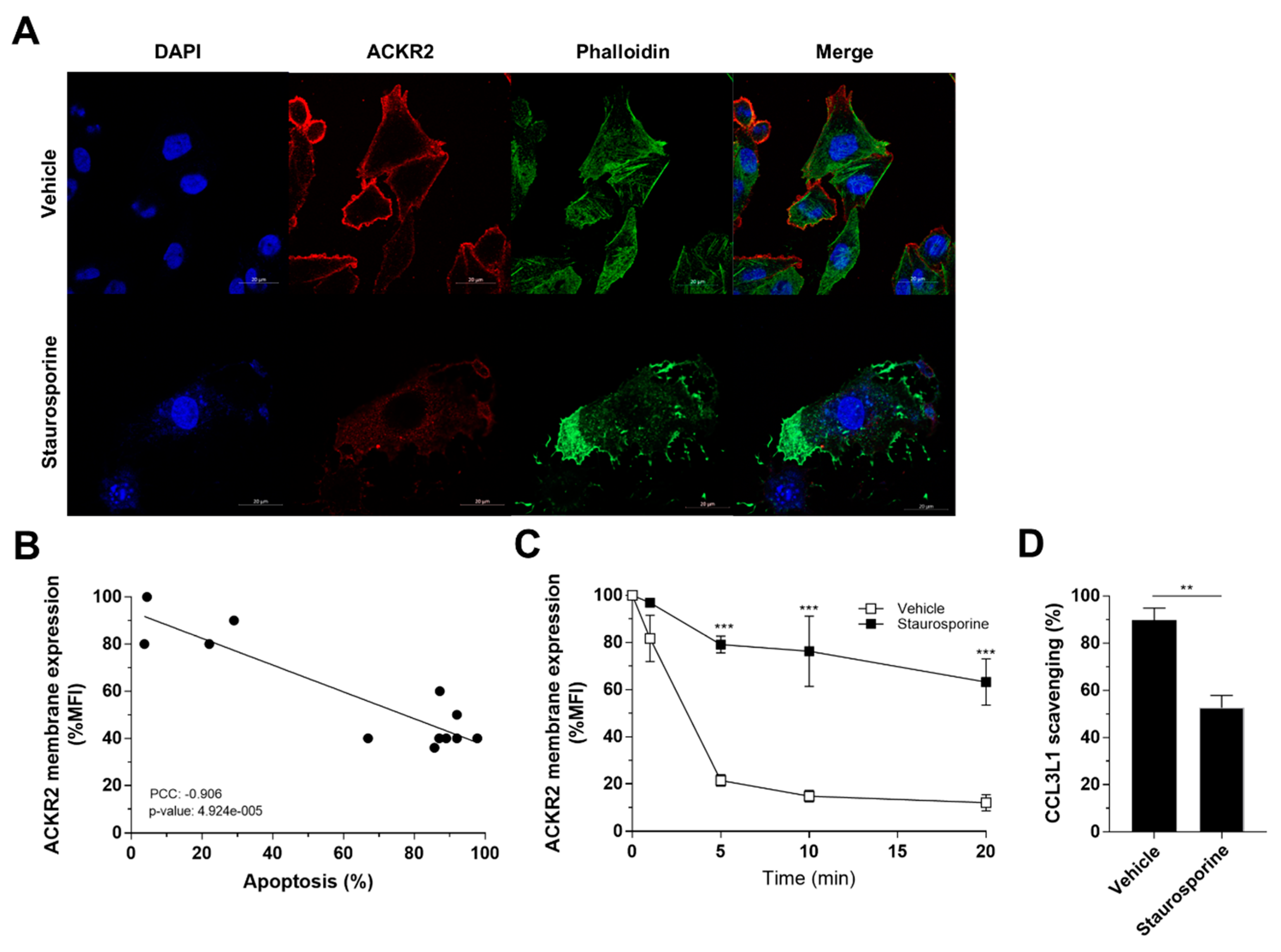
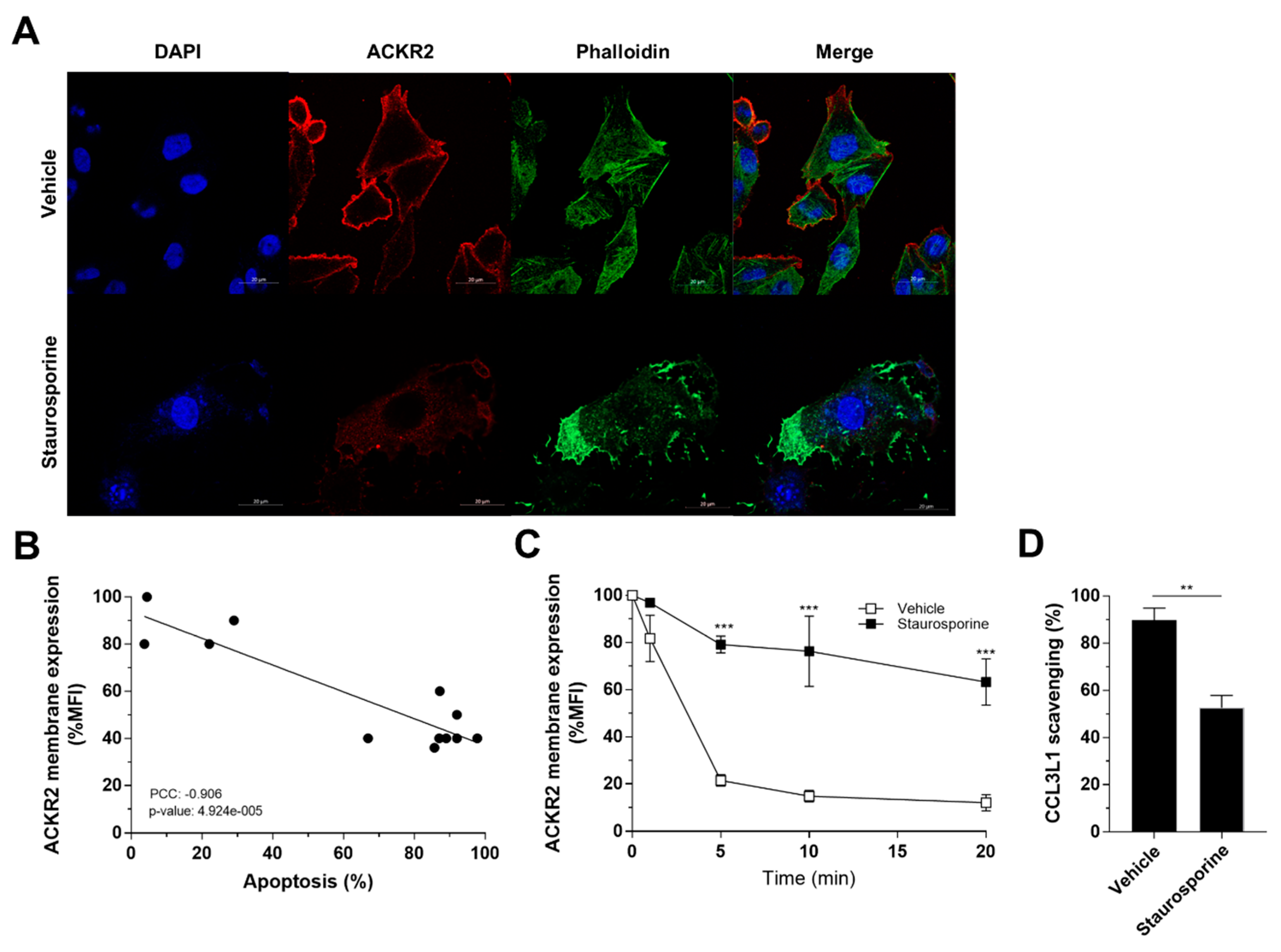
3.6. Apoptosis-Induced Alteration of Actin Dynamics Impairs ACKR2 Internalization and Scavenging
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. Febs J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Lacalle, R.A.; Blanco, R.; Carmona-Rodriguez, L.; Martin-Leal, A.; Mira, E.; Manes, S. Chemokine Receptor Signaling and the Hallmarks of Cancer. Int. Rev. Cell Mol. Biol. 2017, 331, 181–244. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachelerie, F.; Graham, G.J.; Locati, M.; Mantovani, A.; Murphy, P.M.; Nibbs, R.; Rot, A.; Sozzani, S.; Thelen, M. An atypical addition to the chemokine receptor nomenclature: IUPHAR Review 15. Br. J. Pharmacol. 2015, 172, 3945–3949. [Google Scholar] [CrossRef] [Green Version]
- Ulvmar, M.H.; Hub, E.; Rot, A. Atypical chemokine receptors. Exp. Cell Res. 2011, 317, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horuk, R.; Chitnis, C.E.; Darbonne, W.C.; Colby, T.J.; Rybicki, A.; Hadley, T.J.; Miller, L.H. A receptor for the malarial parasite Plasmodium vivax: The erythrocyte chemokine receptor. Science 1993, 261, 1182–1184. [Google Scholar] [CrossRef] [Green Version]
- Nibbs, R.J.; Wylie, S.M.; Yang, J.; Landau, N.R.; Graham, G.J. Cloning and characterization of a novel promiscuous human beta-chemokine receptor D6. J. Biol. Chem. 1997, 272, 32078–32083. [Google Scholar] [CrossRef] [Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef] [Green Version]
- Gosling, J.; Dairaghi, D.J.; Wang, Y.; Hanley, M.; Talbot, D.; Miao, Z.; Schall, T.J. Cutting edge: Identification of a novel chemokine receptor that binds dendritic cell- and T cell-active chemokines including ELC, SLC, and TECK. J. Immunol. 2000, 164, 2851–2856. [Google Scholar] [CrossRef] [Green Version]
- Okinaga, S.; Slattery, D.; Humbles, A.; Zsengeller, Z.; Morteau, O.; Kinrade, M.B.; Brodbeck, R.M.; Krause, J.E.; Choe, H.R.; Gerard, N.P.; et al. C5L2, a nonsignaling C5A binding protein. Biochemistry 2003, 42, 9406–9415. [Google Scholar] [CrossRef] [PubMed]
- Zabel, B.A.; Nakae, S.; Zuniga, L.; Kim, J.Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.; Graham, G.J. Immune regulation by atypical chemokine receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.J. D6/Ackr2. Front. Immunol. 2015, 6, 280. [Google Scholar] [CrossRef] [Green Version]
- Savino, B.; Borroni, E.M.; Torres, N.M.; Proost, P.; Struyf, S.; Mortier, A.; Mantovani, A.; Locati, M.; Bonecchi, R. Recognition versus adaptive up-regulation and degradation of CC chemokines by the chemokine decoy receptor D6 are determined by their N-terminal sequence. J. Biol. Chem. 2009, 284, 26207–26215. [Google Scholar] [CrossRef] [Green Version]
- Nibbs, R.J.; Kriehuber, E.; Ponath, P.D.; Parent, D.; Qin, S.; Campbell, J.D.; Henderson, A.; Kerjaschki, D.; Maurer, D.; Graham, G.J.; et al. The beta-chemokine receptor D6 is expressed by lymphatic endothelium and a subset of vascular tumors. Am. J. Pathol. 2001, 158, 867–877. [Google Scholar] [CrossRef]
- Martinez de la Torre, Y.; Buracchi, C.; Borroni, E.M.; Dupor, J.; Bonecchi, R.; Nebuloni, M.; Pasqualini, F.; Doni, A.; Lauri, E.; Agostinis, C.; et al. Protection against inflammation- and autoantibody-caused fetal loss by the chemokine decoy receptor D6. Proc. Natl. Acad. Sci. USA 2007, 104, 2319–2324. [Google Scholar] [CrossRef] [Green Version]
- Hansell, C.A.; Schiering, C.; Kinstrie, R.; Ford, L.; Bordon, Y.; McInnes, I.B.; Goodyear, C.S.; Nibbs, R.J. Universal expression and dual function of the atypical chemokine receptor D6 on innate-like B cells in mice. Blood 2011, 117, 5413–5424. [Google Scholar] [CrossRef] [Green Version]
- Bazzan, E.; Saetta, M.; Turato, G.; Borroni, E.M.; Cancellieri, C.; Baraldo, S.; Savino, B.; Calabrese, F.; Ballarin, A.; Balestro, E.; et al. Expression of the atypical chemokine receptor D6 in human alveolar macrophages in COPD. Chest 2013, 143, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Bonecchi, R.; Graham, G.J. Atypical Chemokine Receptors and Their Roles in the Resolution of the Inflammatory Response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Vetrano, S.; Borroni, E.M.; Sarukhan, A.; Savino, B.; Bonecchi, R.; Correale, C.; Arena, V.; Fantini, M.; Roncalli, M.; Malesci, A.; et al. The lymphatic system controls intestinal inflammation and inflammation-associated Colon Cancer through the chemokine decoy receptor D6. Gut 2010, 59, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.; Gilchrist, D.S.; King, V.; Ferra, A.; Forrow, S.; Hunter, K.D.; Graham, G.J. The atypical chemokine receptor D6 suppresses the development of chemically induced skin tumors. J. Clin. Investig. 2007, 117, 1884–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savino, B.; Caronni, N.; Anselmo, A.; Pasqualini, F.; Borroni, E.M.; Basso, G.; Celesti, G.; Laghi, L.; Tourlaki, A.; Boneschi, V.; et al. ERK-dependent downregulation of the atypical chemokine receptor D6 drives tumor aggressiveness in Kaposi sarcoma. Cancer Immunol. Res. 2014, 2, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodduluri, S.R.; Mathis, S.; Maturu, P.; Krishnan, E.; Satpathy, S.R.; Chilton, P.M.; Mitchell, T.C.; Lira, S.; Locati, M.; Mantovani, A.; et al. Mast Cell-Dependent CD8(+) T-cell Recruitment Mediates Immune Surveillance of Intestinal Tumors in Apc(Min/+) Mice. Cancer Immunol. Res. 2018, 6, 332–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massara, M.; Bonavita, O.; Savino, B.; Caronni, N.; Mollica Poeta, V.; Sironi, M.; Setten, E.; Recordati, C.; Crisafulli, L.; Ficara, F.; et al. ACKR2 in hematopoietic precursors as a checkpoint of neutrophil release and anti-metastatic activity. Nat. Commun. 2018, 9, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansell, C.A.H.; Fraser, A.R.; Hayes, A.J.; Pingen, M.; Burt, C.L.; Lee, K.M.; Medina-Ruiz, L.; Brownlie, D.; Macleod, M.K.L.; Burgoyne, P.; et al. The Atypical Chemokine Receptor Ackr2 Constrains NK Cell Migratory Activity and Promotes Metastasis. J. Immunol. 2018, 201, 2510–2519. [Google Scholar] [CrossRef]
- Aswad, M.; Assi, S.; Schif-Zuck, S.; Ariel, A. CCL5 Promotes Resolution-Phase Macrophage Reprogramming in Concert with the Atypical Chemokine Receptor D6 and Apoptotic Polymorphonuclear Cells. J. Immunol. 2017, 199, 1393–1404. [Google Scholar] [CrossRef]
- Pashover-Schallinger, E.; Aswad, M.; Schif-Zuck, S.; Shapiro, H.; Singer, P.; Ariel, A. The atypical chemokine receptor D6 controls macrophage efferocytosis and cytokine secretion during the resolution of inflammation. Faseb. J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 3891–3900. [Google Scholar] [CrossRef]
- Weber, M.; Blair, E.; Simpson, C.V.; O’Hara, M.; Blackburn, P.E.; Rot, A.; Graham, G.J.; Nibbs, R.J. The chemokine receptor D6 constitutively traffics to and from the cell surface to internalize and degrade chemokines. Mol. Biol. Cell 2004, 15, 2492–2508. [Google Scholar] [CrossRef] [Green Version]
- Galliera, E.; Jala, V.R.; Trent, J.O.; Bonecchi, R.; Signorelli, P.; Lefkowitz, R.J.; Mantovani, A.; Locati, M.; Haribabu, B. beta-Arrestin-dependent constitutive internalization of the human chemokine decoy receptor D6. J. Biol. Chem. 2004, 279, 25590–25597. [Google Scholar] [CrossRef] [Green Version]
- Bonecchi, R.; Borroni, E.M.; Anselmo, A.; Doni, A.; Savino, B.; Mirolo, M.; Fabbri, M.; Jala, V.R.; Haribabu, B.; Mantovani, A.; et al. Regulation of D6 chemokine scavenging activity by ligand- and Rab11-dependent surface up-regulation. Blood 2008, 112, 493–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroni, E.M.; Cancellieri, C.; Vacchini, A.; Benureau, Y.; Lagane, B.; Bachelerie, F.; Arenzana-Seisdedos, F.; Mizuno, K.; Mantovani, A.; Bonecchi, R.; et al. Beta-Arrestin-Dependent Activation of the Cofilin Pathway Is Required for the Scavenging Activity of the Atypical Chemokine Receptor D6. Sci. Signal. 2013, 6, ra30. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Goldman, R.D. Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 2004, 5, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, S.; Saxena, R.; Chattopadhyay, A. Reorganization of the actin cytoskeleton upon G-protein coupled receptor signaling. Biochim. Biophys. Acta 2011, 1808, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Desnos, C.; Huet, S.; Darchen, F. ‘Should I stay or should I go?’: Myosin V function in organelle trafficking. Biol. Cell Under Auspices Eur. Cell Biol. Organ. 2007, 99, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Roland, J.T.; Bryant, D.M.; Datta, A.; Itzen, A.; Mostov, K.E.; Goldenring, J.R. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc. Natl. Acad. Sci. USA 2011, 108, 2789–2794. [Google Scholar] [CrossRef] [Green Version]
- Lapierre, L.A.; Goldenring, J.R. Interactions of myosin vb with rab11 family members and cargoes traversing the plasma membrane recycling system. Methods Enzymol. 2005, 403, 715–723. [Google Scholar] [CrossRef]
- Lapierre, L.A.; Kumar, R.; Hales, C.M.; Navarre, J.; Bhartur, S.G.; Burnette, J.O.; Provance, D.W., Jr.; Mercer, J.A.; Bahler, M.; Goldenring, J.R. Myosin vb is associated with plasma membrane recycling systems. Mol. Biol. Cell 2001, 12, 1843–1857. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, M.; Worth, D.; Prowse, D.M.; Nikolic, M. Pak1 phosphorylation on t212 affects microtubules in cells undergoing mitosis. Curr. Biol. CB 2002, 12, 1233–1239. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, T.; Bokoch, G.M.; Waterman-Storer, C.M. Regulation of leading edge microtubule and actin dynamics downstream of Rac1. J. Cell Biol. 2003, 161, 845–851. [Google Scholar] [CrossRef]
- Prudent, R.; Vassal-Stermann, E.; Nguyen, C.H.; Pillet, C.; Martinez, A.; Prunier, C.; Barette, C.; Soleilhac, E.; Filhol, O.; Beghin, A.; et al. Pharmacological inhibition of LIM kinase stabilizes microtubules and inhibits neoplastic growth. Cancer Res. 2012, 72, 4429–4439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, M.; Di Meglio, S.; Gagliani, M.C.; Consonni, E.; Molteni, R.; Bender, J.R.; Tacchetti, C.; Pardi, R. Dynamic partitioning into lipid rafts controls the endo-exocytic cycle of the alphaL/beta2 integrin, LFA-1, during leukocyte chemotaxis. Mol. Biol. Cell 2005, 16, 5793–5803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, G.H.; Lapierre, L.A.; Goldenring, J.R.; Sai, J.; Richmond, A. Rab11-family interacting protein 2 and myosin Vb are required for CXCR2 recycling and receptor-mediated chemotaxis. Mol. Biol. Cell 2004, 15, 2456–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, K.; Fujiwara, S.; Watanabe, T.; Kondo, H.; Kiuchi, T.; Sato, M.; Mizuno, K. LIM kinase has a dual role in regulating lamellipodium extension by decelerating the rate of actin retrograde flow and the rate of actin polymerization. J. Biol. Chem. 2011, 286, 36340–36351. [Google Scholar] [CrossRef] [Green Version]
- Fra, A.M.; Locati, M.; Otero, K.; Sironi, M.; Signorelli, P.; Massardi, M.L.; Gobbi, M.; Vecchi, A.; Sozzani, S.; Mantovani, A. Cutting edge: Scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J. Immunol. 2003, 170, 2279–2282. [Google Scholar] [CrossRef]
- Mostowy, S.; Shenoy, A.R. The cytoskeleton in cell-autonomous immunity: Structural determinants of host defence. Nat. Rev. Immunol. 2015, 15, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef]
- Vacchini, A.; Locati, M.; Borroni, E.M. Overview and potential unifying themes of the atypical chemokine receptor family. J. Leukoc. Biol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, K.; Li, R.; Soo, P.; Suryadinata, R.; Sarcevic, B.; Valova, V.A.; Graham, M.E.; Robinson, P.J.; Bernard, O. The phosphorylation of p25/TPPP by LIM kinase 1 inhibits its ability to assemble microtubules. Exp. Cell Res. 2007, 313, 4091–4106. [Google Scholar] [CrossRef]
- Trybus, K.M. Myosin V from head to tail. Cell. Mol. Life Sci. Cmls 2008, 65, 1378–1389. [Google Scholar] [CrossRef]
- Legler, D.F.; Thelen, M. New insights in chemokine signaling. F1000Research 2018, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innamorati, G.; Le Gouill, C.; Balamotis, M.; Birnbaumer, M. The long and the short cycle. Alternative intracellular routes for trafficking of G-protein-coupled receptors. J. Biol. Chem. 2001, 276, 13096–13103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, C.V.; Morrow, V.; Milasta, S.; Comerford, I.; Milligan, G.; Graham, G.J.; Isaacs, N.W.; Nibbs, R.J. Multiple roles for the C-terminal tail of the chemokine scavenger D6. J. Biol. Chem. 2008, 283, 7972–7982. [Google Scholar] [CrossRef] [Green Version]
- Franklin-Tong, V.E.; Gourlay, C.W. A role for actin in regulating apoptosis/programmed cell death: Evidence spanning yeast, plants and animals. Biochem. J. 2008, 413, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Gourlay, C.W.; Ayscough, K.R. The actin cytoskeleton: A key regulator of apoptosis and ageing? Nat. Rev. Mol. Cell Biol. 2005, 6, 583–589. [Google Scholar] [CrossRef]
- Povea-Cabello, S.; Oropesa-Avila, M.; de la Cruz-Ojeda, P.; Villanueva-Paz, M.; de la Mata, M.; Suarez-Rivero, J.M.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Cotan, D.; Ybot-Gonzalez, P.; et al. Dynamic Reorganization of the Cytoskeleton during Apoptosis: The Two Coffins Hypothesis. Int. J. Mol. Sci. 2017, 18, 2393. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vacchini, A.; Cancellieri, C.; Milanesi, S.; Badanai, S.; Savino, B.; Bifari, F.; Locati, M.; Bonecchi, R.; Borroni, E.M. Control of Cytoskeletal Dynamics by β-Arrestin1/Myosin Vb Signaling Regulates Endosomal Sorting and Scavenging Activity of the Atypical Chemokine Receptor ACKR2. Vaccines 2020, 8, 542. https://doi.org/10.3390/vaccines8030542
Vacchini A, Cancellieri C, Milanesi S, Badanai S, Savino B, Bifari F, Locati M, Bonecchi R, Borroni EM. Control of Cytoskeletal Dynamics by β-Arrestin1/Myosin Vb Signaling Regulates Endosomal Sorting and Scavenging Activity of the Atypical Chemokine Receptor ACKR2. Vaccines. 2020; 8(3):542. https://doi.org/10.3390/vaccines8030542
Chicago/Turabian StyleVacchini, Alessandro, Cinzia Cancellieri, Samantha Milanesi, Sabrina Badanai, Benedetta Savino, Francesco Bifari, Massimo Locati, Raffaella Bonecchi, and Elena Monica Borroni. 2020. "Control of Cytoskeletal Dynamics by β-Arrestin1/Myosin Vb Signaling Regulates Endosomal Sorting and Scavenging Activity of the Atypical Chemokine Receptor ACKR2" Vaccines 8, no. 3: 542. https://doi.org/10.3390/vaccines8030542
APA StyleVacchini, A., Cancellieri, C., Milanesi, S., Badanai, S., Savino, B., Bifari, F., Locati, M., Bonecchi, R., & Borroni, E. M. (2020). Control of Cytoskeletal Dynamics by β-Arrestin1/Myosin Vb Signaling Regulates Endosomal Sorting and Scavenging Activity of the Atypical Chemokine Receptor ACKR2. Vaccines, 8(3), 542. https://doi.org/10.3390/vaccines8030542