Whole Genome In-Silico Analysis of South African G1P[8] Rotavirus Strains before and after Vaccine Introduction over a Period of 14 Years


 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. Strain Description
2.3. Extraction and Purification of Double-Stranded RNA
2.4. Complementary DNA (Cdna) Synthesis
2.5. DNA Library Preparation and Whole-Genome Sequencing
2.6. Genome Assembly
2.7. Generation of Whole-Genome Constellations
2.8. Phylogenetic Analyses
2.9. Selection Pressure and Recombination Analysis
2.10. Protein Modeling
2.11. In Silico Analysis of Effect of Mutation(s) on Protein Stability
3. Results
3.1. Whole-Genome Constellation Determination
3.2. Phylogenetic Analyses
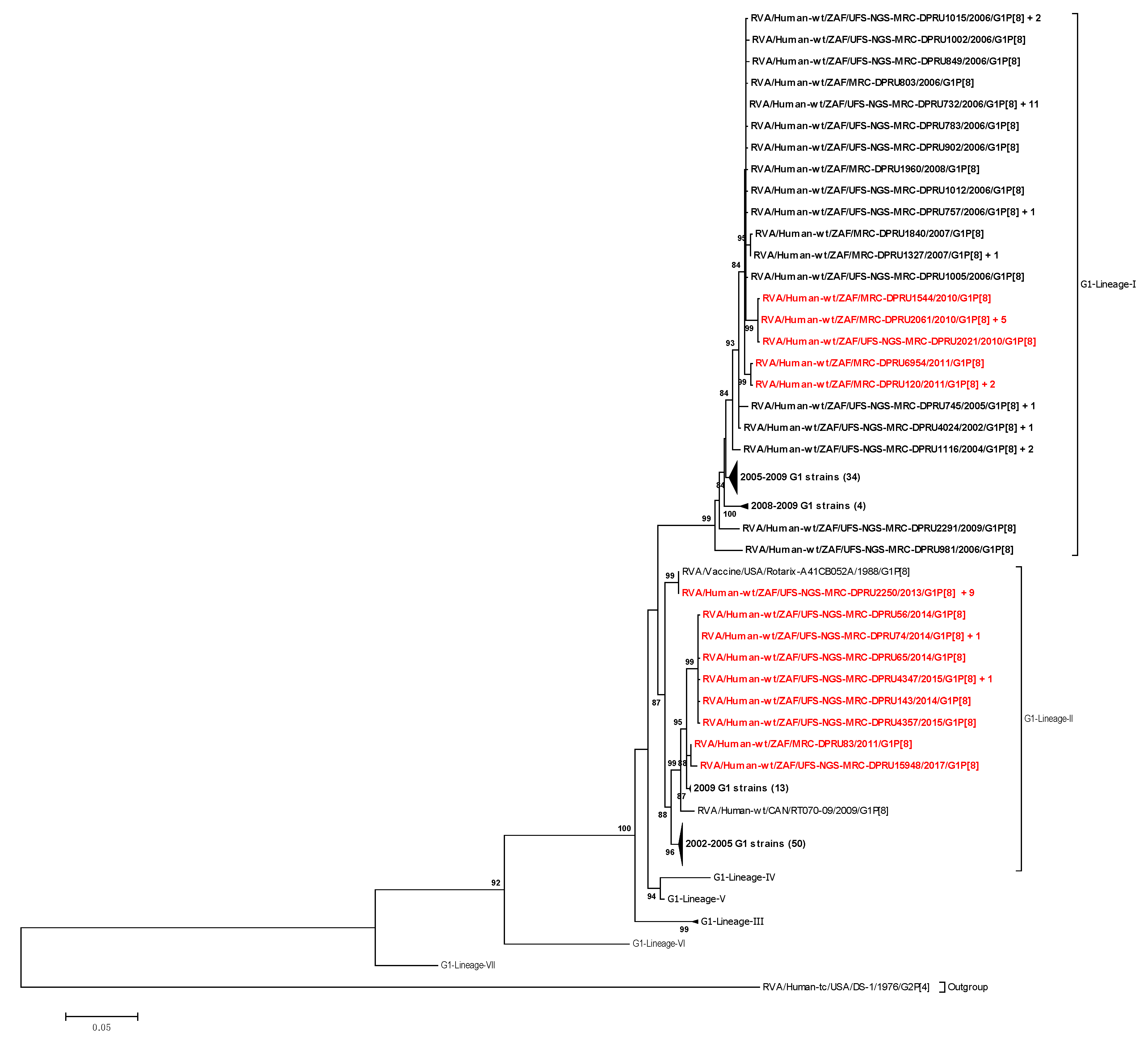
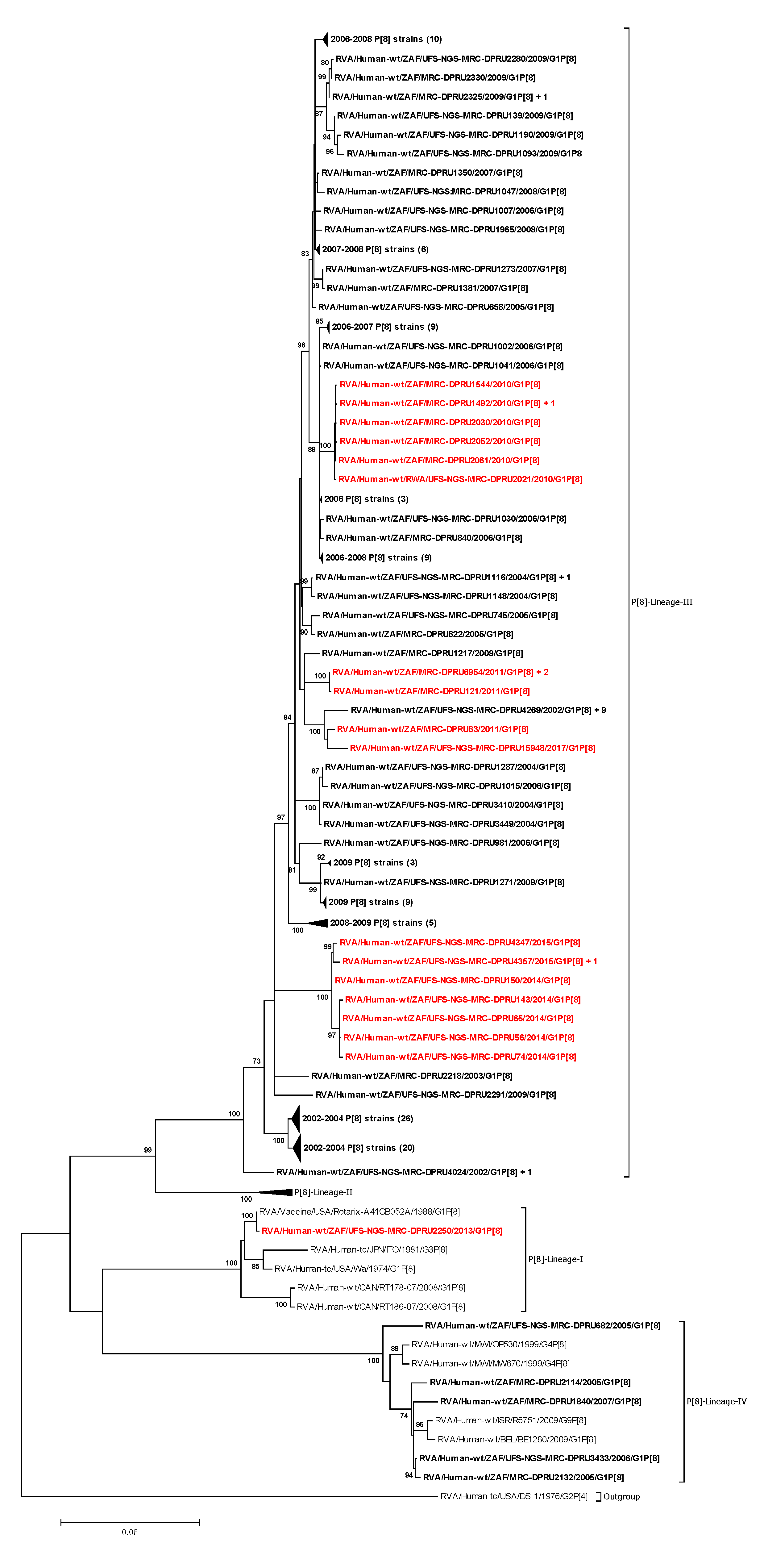
3.2.1. Phylogenetic Analyses of VP7 and VP4
3.2.2. Phylogenetic Analysis of VP1–VP3, VP6, and NSP1–NSP5
3.3. Protein Modeling and Amino Acid Analysis
3.3.1. Comparative Analysis of Neutralizing Antigenic VP7 Epitopes of South African G1 Strains and Rotarix® Strains
3.3.2. Comparative Analysis of VP7 Cytotoxic T Lymphocyte Epitopes of South Africa With Rotarix® Vaccine Strain
3.3.3. Comparative Analysis of Neutralizing Antigenic Epitopes in VP4 Genes of South African P[8] Strains and Rotarix® Vaccine Strain
3.3.4. Analysis of the VP4 and VP6 Non-Neutralizing Regions
3.3.5. Analysis of Amino Acid Differences in VP1–VP3 and NSP1–NSP5 Amino Acid Sequences
3.3.6. Analyses of Selection Pressure and Recombination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estes, M.K.; Kapikian, A. Rotaviruses. In Fields Virology; Knipe, D., Griffin, D., Lamb, R., Martin, M., Roizman, B., Straus, S., Eds.; Wolters Kluwer Health; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 1917–1975. [Google Scholar]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group a rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Dóró, R.; László, B.; Martella, V.; Leshem, E.; Gentsch, J.; Parashar, U.; Bányai, K. Review of global rotavirus strain prevalence data from six years post vaccine licensure surveillance: Is there evidence of strain selection from vaccine pressure? Infect. Genet. Evol. 2014, 28, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, C.D. Genetic and antigenic diversity of human rotaviruses: Potential impact on vaccination programs. J. Infect. Dis. 2010, 202, S43–S48. [Google Scholar] [CrossRef]
- Arista, S.; Giammanco, G.M.; De Grazia, S.; Ramirez, S.; Biundo, C.L.; Colomba, C.; Cascio, A.; Martella, V. Heterogeneity and temporal dynamics of evolution of G1 human rotaviruses in a settled population. J. Virol. 2006, 80, 10724–10733. [Google Scholar] [CrossRef] [Green Version]
- Bányai, K.; Gentsch, J.R.; Martella, V.; Bogdán, Á.; Havasi, V.; Kisfali, P.; Szabó, A.; Mihály, I.; Molnár, P.; Melegh, B.; et al. Trends in the epidemiology of human G1P [8] rotaviruses: A Hungarian study. J. Infect. Dis. 2009, 200 (Suppl. 1), S222–S227. [Google Scholar] [CrossRef]
- Le, V.P.; Chung, Y.C.; Kim, K.; Chung, S.I.; Lim, I.; Kim, W. Genetic variation of prevalent G1P [8] human rotaviruses in South Korea. J. Med. Virol. 2010, 82, 886–896. [Google Scholar] [CrossRef]
- Bucardo, F.; Rippinger, C.M.; Svensson, L.; Patton, J.T. Vaccine-derived NSP2 segment in rotaviruses from vaccinated children with gastroenteritis in Nicaragua. Infect. Genet. Evol. 2012, 12, 1282–1294. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.K.; Jheong, W.H.; Lee, S.G.; Park, C.J.; Jung, K.H.; Paik, S.Y. Full genomic analysis of a human rotavirus G1P [8] strain isolated in South Korea. J. Med. Virol. 2013, 85, 157–170. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Prequalifies New Rotavirus Vaccine. Available online: https://www.who.int/medicines/news/2018/prequalified_new-rotavirus_vaccine/en/ (accessed on 21 January 2020).
- Bernstein, D.I.; Sack, D.A.; Rothstein, E.; Reisinger, K.; Smith, V.E.; O’Sullivan, D.; Spriggs, D.R.; Ward, R.L. Efficacy of live, attenuated, human rotavirus vaccine 89–12 in infants: A randomised placebo-controlled trial. Lancet 1999, 354, 287–290. [Google Scholar] [CrossRef]
- Ciarlet, M.; Schödel, F. Development of a rotavirus vaccine: Clinical safety, immunogenicity, and efficacy of the pentavalent rotavirus vaccine, RotaTeq®. Vaccine 2009, 27, G72–G81. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, N.; Rongsen-Chandola, T.; Bavdekar, A.; John, J.; Antony, K.; Taneja, S.; Goyal, N.; Kawade, A.; Kang, G.; Rathore, S.S.; et al. Efficacy of a monovalent human-bovine (116E) rotavirus vaccine in Indian infants: A randomised, double-blind, placebo-controlled trial. Lancet 2014, 383, 2136–2143. [Google Scholar] [CrossRef] [Green Version]
- Zade, J.K.; Kulkarni, P.S.; Desai, S.A.; Sabale, R.N.; Naik, S.P.; Dhere, R.M. Bovine rotavirus pentavalent vaccine development in India. Vaccine 2014, 32, A124–A128. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Rotavirus Vaccination, Meeting of the immunization Strategic Advisory Group of Experts, April 2009. Wkly. Epidemiol. Rec. 2009, 84, 220–236. [Google Scholar]
- Msimang, V.M.; Page, N.; Groome, M.J.; Moyes, J.; Cortese, M.M.; Seheri, M.; Kahn, K.; Chagan, M.; Madhi, S.A.; Cohen, C. Impact of rotavirus vaccine on childhood diarrheal hospitalization after introduction into the South African public immunization program. Pediatr. Infect. Dis. J. 2013, 32, 1359–1364. [Google Scholar] [CrossRef]
- Page, N.A.; Seheri, L.M.; Groome, M.J.; Moyes, J.; Walaza, S.; Mphahlele, J.; Kahn, K.; Kapongo, C.N.; Zar, H.J.; Tempia, S.; et al. Temporal association of rotavirus vaccination and genotype circulation in South Africa: Observations from 2002 to 2014. Vaccine 2018, 36, 7231–7237. [Google Scholar] [CrossRef]
- Steele, A.D.; Peenze, I.; De Beer, M.C.; Pager, C.T.; Yeats, J.; Potgieter, N.; Ramsaroop, U.; Page, N.A.; Mitchell, J.O.; Geyer, A.; et al. Anticipating rotavirus vaccines: Epidemiology and surveillance of rotavirus in South Africa. Vaccine 2003, 21, 354–360. [Google Scholar] [CrossRef]
- Hoxie, I.; Dennehy, J.J. Intragenic recombination influences rotavirus diversity and evolution. Virus Evol. 2020, 6, vez059. [Google Scholar] [CrossRef]
- Esona, M.D.; Roy, S.; Rungsrisuriyachai, K.; Gautam, R.; Hermelijn, S.; Rey-Benito, G.; Bowen, M.D. Molecular characterization of a human G20P [28] rotavirus a strain with multiple genes related to bat rotaviruses. Infect. Genet. Evol. 2018, 57, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoa-Tran, T.N.; Nakagomi, T.; Vu, H.M.; Nguyen, T.T.T.; Takemura, T.; Hasebe, F.; Dao, A.T.H.; Anh, P.H.Q.; Nguyen, A.T.; Dang, A.D.; et al. Detection of three independently-generated DS-1-like G9P [8] reassortant rotavirus A strains during the G9P [8] dominance in Vietnam, 2016–2018. Infect. Genet. Evol. 2020, 80, 104194. [Google Scholar] [CrossRef] [PubMed]
- Maringa, W.M.; Mwangi, P.N.; Simwaka, J.; Mpabalwani, E.M.; Mwenda, J.M.; Peenze, I.; Esona, M.D.; Mphahlele, M.J.; Seheri, M.L.; Nyaga, M.M. Molecular Characterisation of a Rare Reassortant Porcine-Like G5P[6] Rotavirus Strain Detected in an Unvaccinated Child in Kasama, Zambia. Pathogens 2020, 9, 663. [Google Scholar] [CrossRef]
- Shah, M.P.; Tate, J.E.; Mwenda, J.M.; Steele, A.D.; Parashar, U.D. Estimated reductions in hospitalizations and deaths from childhood diarrhea following implementation of rotavirus vaccination in Africa. Expert Rev. Vaccines 2017, 16, 987–995. [Google Scholar] [CrossRef]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. 655 GenBank. Nucleic Acids Res 2015, 43, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; Van Dijk, A.A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 277, pp. 396–404. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the FoldX forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.T.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [Green Version]
- Maiorov, V.N.; Crippen, G.M. Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Morozova, O.V.; Sashina, T.A.; Fomina, S.G.; Novikova, N.A. Comparative characteristics of the VP7 and VP4 antigenic epitopes of the rotaviruses circulating in Russia (Nizhny Novgorod) and the Rotarix and RotaTeq vaccines. Arch. Virol. 2015, 160, 1693–1703. [Google Scholar] [CrossRef]
- Dormitzer, P.R.; Sun, Z.Y.J.; Wagner, G.; Harrison, S.C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 2002, 21, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. Bioinform. Genet. 2003, 317, 289. [Google Scholar]
- Tosser, G.; Labbe, M.; Bremont, M.; Cohen, J. Expression of the major capsid protein VP6 of group C rotavirus and synthesis of chimeric single-shelled particles by using recombinant baculoviruses. J. Virol. 1992, 66, 5825–5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.; Matthijnssens, J.; Saiada, F.; Hassan, Z.; Heylen, E.; Azim, T.; Van Ranst, M. Complete genomic analysis of a Bangladeshi G1P [8] rotavirus strain detected in 2003 reveals a close evolutionary relationship with contemporary human Wa-like strains. Infect. Genet. Evol. 2010, 10, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Bányai, K.; László, B.; Duque, J.; Steele, A.D.; Nelson, E.A.S.; Gentsch, J.R.; Parashar, U.D. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: Insights for understanding the impact of rotavirus vaccination programs. Vaccine 2012, 30, A122–A130. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Ghosh, S.; Wang, Y.H.; Zhou, X.; Zhou, D.J.; Kobayashi, N. Whole genomic analysis of human G1P [8] rotavirus strains from different age groups in China. Viruses 2012, 4, 1289–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, F.S.; Junior, E.S.; Guerra, S.D.F.D.S.; Lobo, P.S.; Junior, E.P.; Lima, A.B.F.; Vinente, C.B.G.; Chagas, E.H.N.; Justino, M.C.A.; Linhares, A.D.C.; et al. G1P [8] Rotavirus in children with severe diarrhea in the post-vaccine introduction era in Brazil: Evidence of reassortments and structural modifications of the antigenic VP7 and VP4 regions. Infect. Genet. Evol. 2019, 69, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kobayashi, N. Whole-genomic analysis of rotavirus strains: Current status and future prospects. Future Microbiol. 2011, 6, 1049–1065. [Google Scholar] [CrossRef]
- da Silva, M.F.M.; Rose, T.L.; Gómez, M.M.; Carvalho-Costa, F.A.; Fialho, A.M.; de Assis, R.M.S.; de Andrade, J.D.S.R.; de Mello Volotão, E.; Leite, J.P.G. G1P [8] species A rotavirus over 27 years–pre-and post-vaccination eras–in Brazil: Full genomic constellation analysis and no evidence for selection pressure by Rotarix® vaccine. Infect. Genet. Evol. 2015, 30, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Dulgheroff, A.C.B.; Silva, G.A.V.D.; Naveca, F.G.; Oliveira, A.G.D.; Domingues, A.L.D.S. Diversity of group A rotavirus genes detected in the Triângulo Mineiro region, Minas Gerais, Brazil. Braz. J. Microbiol. 2016, 47, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Almeida, T.N.V.; de Sousa, T.T.; da Silva, R.A.; Fiaccadori, F.S.; Souza, M.; Badr, K.R.; de Paula Cardoso, D.D.D. Phylogenetic analysis of G1P [8] and G12P [8] rotavirus A samples obtained in the pre-and post-vaccine periods, and molecular modeling of VP4 and VP7 proteins. Acta Trop. 2017, 173, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, N.A.; Gondwe, J.S.; Graham, S.M.; Thindwa, B.D.M.; Dove, W.; Broadhead, R.L.; Molyneux, M.E.; Hart, C.A. Rotavirus strain diversity in Blantyre, Malawi, from 1997 to 1999. J. Clin. Microbiol. 2001, 39, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatte, V.S.; Chitambar, D.S.D. Intragenotypic diversity in the VP4 encoding genes of rotavirus strains circulating in adolescent and adult cases of acute gastroenteritis in Pune, Western India: 1993 to 1996 and 2004 to 2007. J. Gen. Mol. Virol. 2011, 3, 53–62. [Google Scholar]
- Gouvea, V.; Lima, R.C.; Linhares, R.E.; Clark, H.F.; Nosawa, C.M.; Santos, N. Identification of two lineages (WA-like and F45-like) within the major rotavirus genotype P [8]. Virus Res. 1999, 59, 141–147. [Google Scholar] [CrossRef]
- Ansaldi, F.; Pastorino, B.; Valle, L.; Durando, P.; Sticchi, L.; Tucci, P.; Biasci, P.; Lai, P.; Gasparini, R.; Icardi, G. Molecular characterization of a new variant of rotavirus P [8] G9 predominant in a sentinel-based survey in central Italy. J. Clin. Microbiol. 2007, 45, 1011–1015. [Google Scholar] [CrossRef] [Green Version]
- Espinola, E.E.; Amarilla, A.; Arbiza, J.; Parra, G.I. Sequence and phylogenetic analysis of the VP4 gene of human rotaviruses isolated in Paraguay. Arch. Virol. 2008, 153, 1067–1073. [Google Scholar] [CrossRef]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic analyses reveal differences in the VP7 and VP4 antigenic epitopes between human rotaviruses circulating in Belgium and rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Fredj, M.B.H.; BenHamida-Rebaï, M.; Heylen, E.; Zeller, M.; Moussa, A.; Kacem, S.; Van Ranst, M.; Matthijnssens, J.; Trabelsi, A. Sequence and phylogenetic analyses of human rotavirus strains: Comparison of VP7 and VP8∗ antigenic epitopes between Tunisian and vaccine strains before national rotavirus vaccine introduction. Infect. Genet. Evol. 2013, 18, 132–144. [Google Scholar] [CrossRef]
- Caust, J.; Dyall-Smith, M.L.; Lazdins, I.; Holmes, I.H. Glycosylation, an important modifier of rotavirus antigenicity. Arch. Virol. 1987, 96, 123–134. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Gao, Y.; Zhang, J.; Zhenirovskyy, M.; Alexov, E. Predicting folding free energy changes upon single point mutations. Bioinformatics 2012, 28, 664–671. [Google Scholar] [CrossRef]
- Ogden, K.M.; Snyder, M.J.; Dennis, A.F.; Patton, J.T. Predicted structure and domain organization of rotavirus capping enzyme and innate immune antagonist VP3. J. Virol. 2014, 88, 9072–9085. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Segments/Nucleotide Identity Values in Percentage | VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Comparison between pre- and post-vaccine G1P[8] strains | 92.1–100 | 87.8–99.5 | 89.0–99.4 | 93.7–99.0 | 92.9–99.6 | 89.4–99.1 | 83.5–99.5 | 89.1–99.7 | 94.2–100 | 91.0–100 | 92.2–99.8 |
| Comparison between pre-vaccine G1P[8] strains and Rotarix® strain | 92.7–100 | 89.6–91.1 | 88.2–98.7 | 94.5–99.1 | 93.1–99.0 | 91.2–98.6 | 83.6–100 | 88.3–90.9 | 95.3–100 | 91.4–98.9 | 92.2–98.8 |
| Comparison between post-vaccine strains and Rotarix® strain | 93.3–100 | 89.9–99.9 | 88.9–100 | 94.5–100 | 93.0–100 | 90.9–99.9 | 84.0–99.9 | 89.7–100 | 97.1–100 | 91.0–100 | 92.9–100 |
| Strain Used for the Protein Modeling | Mutation | No. of Post-Vaccine Strain(s) with the Mutation | Region | Amino Acid Property Change | Superposition Value (RMSD) | Free Energy Change (kcal/mol) | Possible Effect |
|---|---|---|---|---|---|---|---|
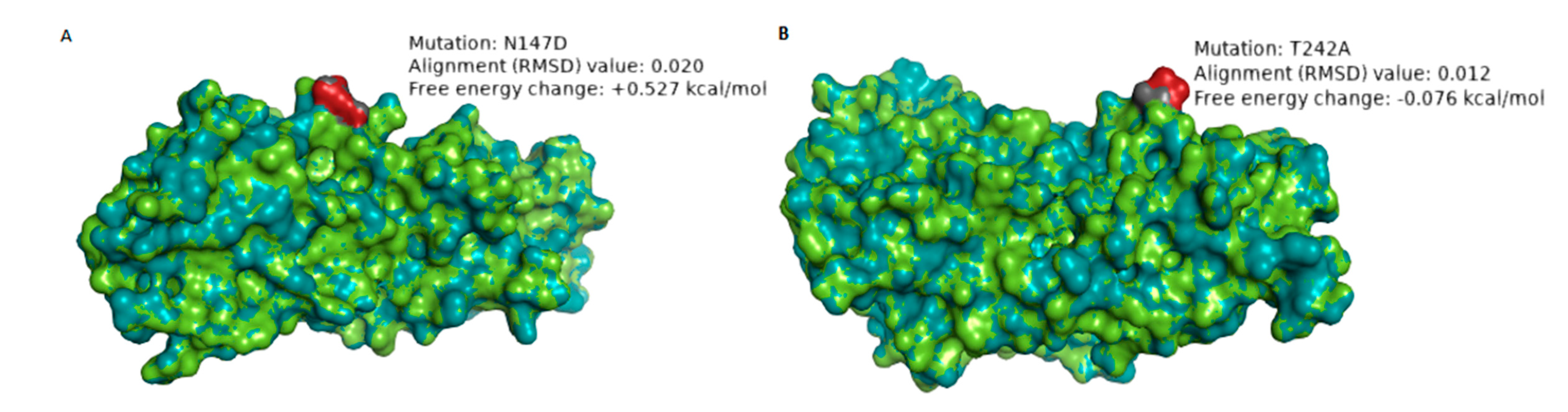
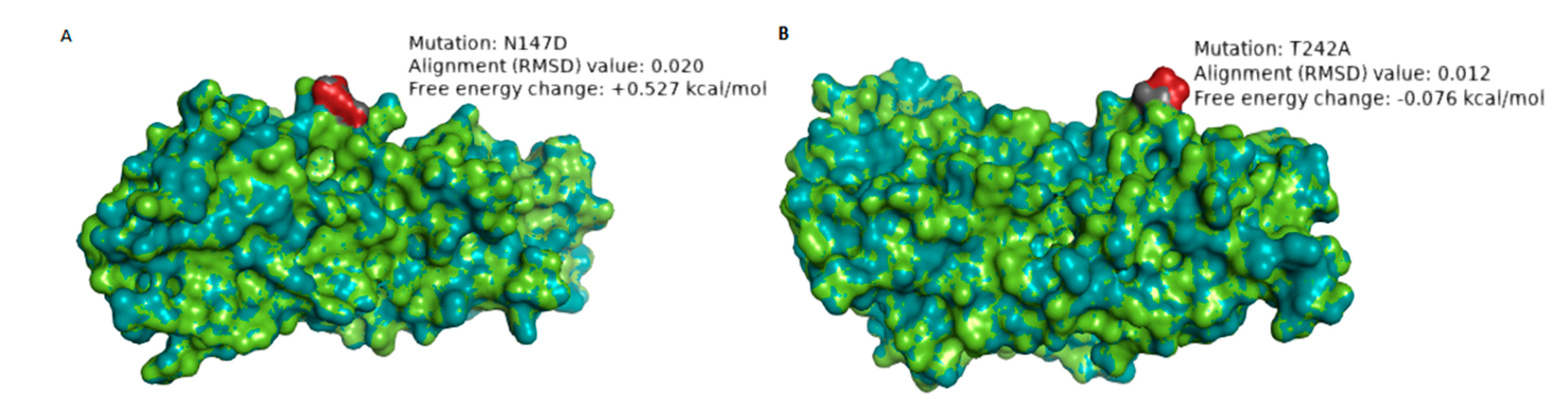
| RVA/Human-wt/ZAF/UFS-NGS-MRC-DPRU4357/2015/G1P[8] | N147D | 8 | 7–2 epitope | Hydrophilic to hydrophilic; Neutral to negative charge | 0.020 Å | +0.527 | The change in charge may alter the biochemical properties of the epitope. The mutation significantly destabilizes the structure of the protein. |
| RVA/Human-wt/ZAF/MRC-DPRU1544/2010/G1P[8] | T242A | 1 | 7–1b epitope | Hydrophilic to hydrophobic; Neutral to Neutral charge | 0.012 Å | −0.076 | The change in polarity may alter the physicochemical property of the epitope |
| Strain Used for Protein Modeling | Mutation | No. of Strain(s) with the Mutation | Region | Amino ACID Property Change | Superimposition Value (RMSD) | Free Energy Change (kcal/mol) | Possible Effect |
|---|---|---|---|---|---|---|---|
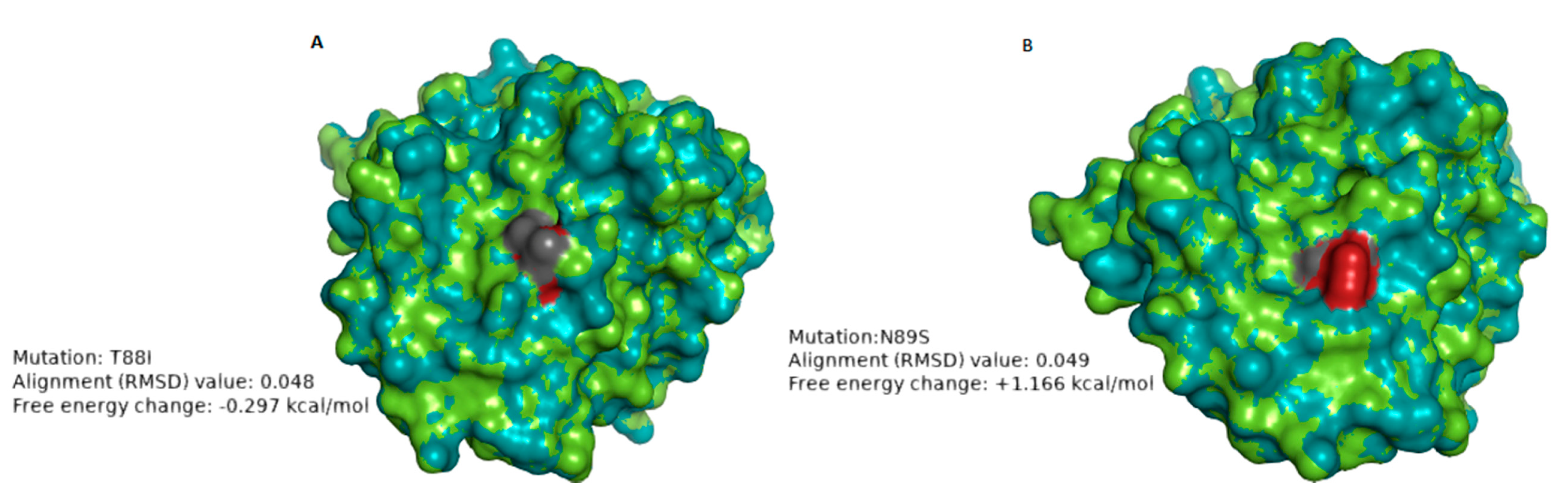
| RVA/Human-wt/ZAF/UFS-NGS-MRC-DPRU74/2014/G1P[8] | T88I | 1 | 8–4 epitope | Hydrophilic to hydrophobic; Neutral charge to neutral charge | 0.048 Å | −0.297 | The change in polarity may alter the physicochemical properties of the protein. No significant impact on the stability of the protein structure |
| RVA/Human-wt/ZAF/UFS-NGS-MRC-DPRU83/2011/G1P[8] | N89S | 1 | 8–4 epitope | Hydrophilic to hydrophilic; Neutral charge to Neutral charge | 0.049 Å | +1.166 | The loss of the glycosylation site may alter the chemical properties of the protein. The mutation destabilized the protein structure. |
| Method | Amino Acid Sites in the Gene Segments under Positive Selection | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| VP1 | VP2 | VP3 | VP4 | VP6 | VP7 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 | |
| MEME | 2, 3, 5, 89, 243, 357, 733, 909, 1082 | 583 | 7, 118, 137, 245, 310, 728 | 23, 28, 44, 90, 169, 201, 576, 578, 668, 773, 774, 775 | 85, 238 | - | 14, 32, 90, 132, 154, 175, 181, 225, 253, 261, 267, 293, 392, 396, 398, 404, 405, 416, 425 | 55, 258, 315 | 204 | 25, 168 | 3 |
| FUBAR | 67, 357 | 12, 36 | 7, 245 | - | - | - | - | 75 | 308 | - | - |
| FEL | 3 | - | 7, 245 | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwangi, P.N.; Mogotsi, M.T.; Seheri, M.L.; Mphahlele, M.J.; Peenze, I.; Esona, M.D.; Kumwenda, B.; Steele, A.D.; Kirkwood, C.D.; Ndze, V.N.; et al. Whole Genome In-Silico Analysis of South African G1P[8] Rotavirus Strains before and after Vaccine Introduction over a Period of 14 Years. Vaccines 2020, 8, 609. https://doi.org/10.3390/vaccines8040609
Mwangi PN, Mogotsi MT, Seheri ML, Mphahlele MJ, Peenze I, Esona MD, Kumwenda B, Steele AD, Kirkwood CD, Ndze VN, et al. Whole Genome In-Silico Analysis of South African G1P[8] Rotavirus Strains before and after Vaccine Introduction over a Period of 14 Years. Vaccines. 2020; 8(4):609. https://doi.org/10.3390/vaccines8040609
Chicago/Turabian StyleMwangi, Peter N., Milton T. Mogotsi, Mapaseka L. Seheri, M. Jeffrey Mphahlele, Ina Peenze, Mathew D. Esona, Benjamin Kumwenda, A. Duncan Steele, Carl D. Kirkwood, Valantine N. Ndze, and et al. 2020. "Whole Genome In-Silico Analysis of South African G1P[8] Rotavirus Strains before and after Vaccine Introduction over a Period of 14 Years" Vaccines 8, no. 4: 609. https://doi.org/10.3390/vaccines8040609
APA StyleMwangi, P. N., Mogotsi, M. T., Seheri, M. L., Mphahlele, M. J., Peenze, I., Esona, M. D., Kumwenda, B., Steele, A. D., Kirkwood, C. D., Ndze, V. N., Dennis, F. E., Jere, K. C., & Nyaga, M. M. (2020). Whole Genome In-Silico Analysis of South African G1P[8] Rotavirus Strains before and after Vaccine Introduction over a Period of 14 Years. Vaccines, 8(4), 609. https://doi.org/10.3390/vaccines8040609