B and T Cell Immunity in Tissues and Across the Ages


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune Cells in Peripheral Blood: Main B- and T-cell Populations
2.1. B Cells: A Complex Population Composed of Various Subsets with Diverse Roles in Immunity
2.1.1. Plasmablasts (PBs)
2.1.2. Long-Lived Plasma Cells (LLPC)
2.1.3. Memory B (BM) Cells
2.1.4. Atypical B Cells
2.2. T Cells in Peripheral Blood: Effector and Memory Cells
2.3. T Follicular Helper (TFH) Cells
3. Immune Cells in the Gut
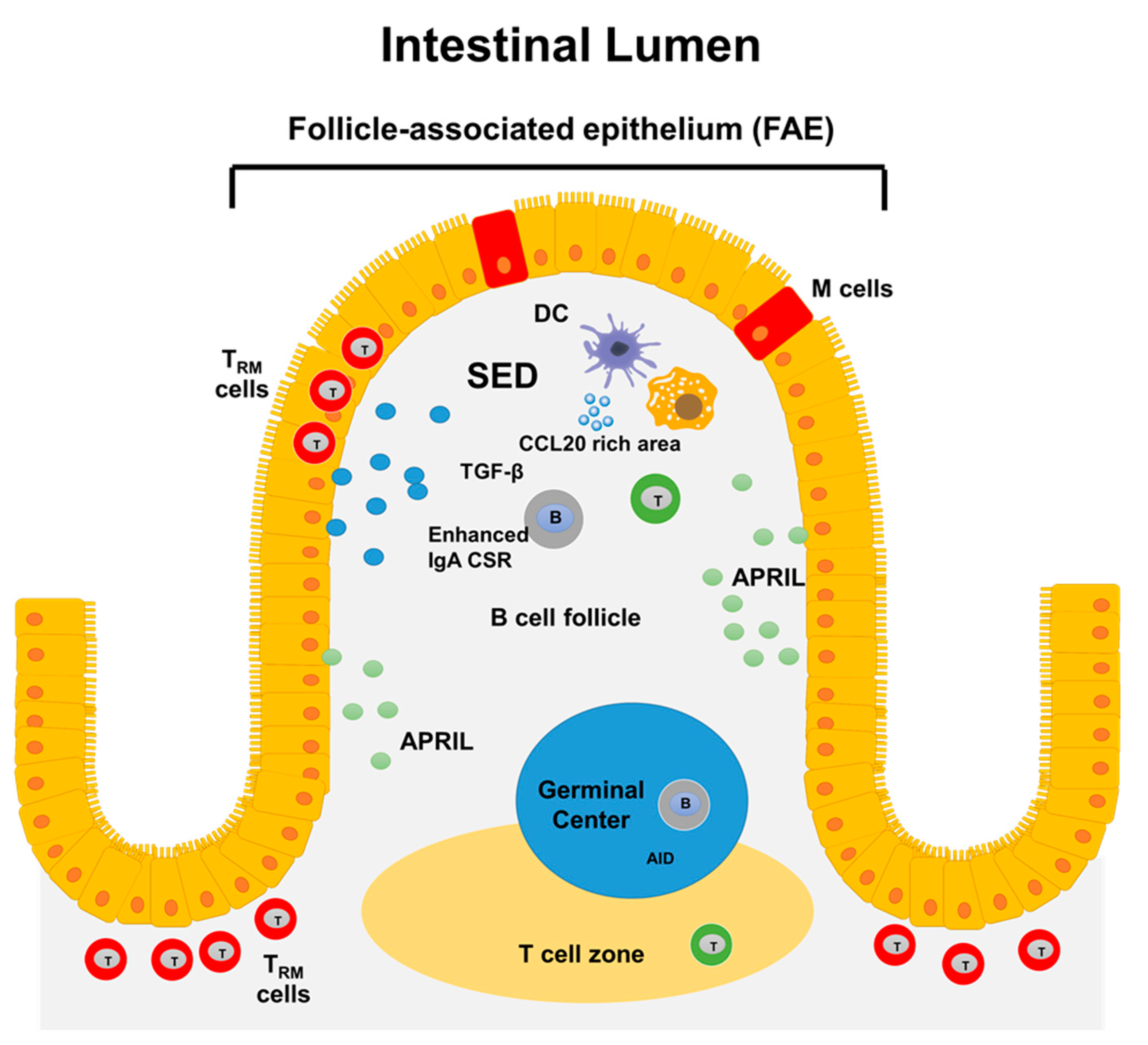
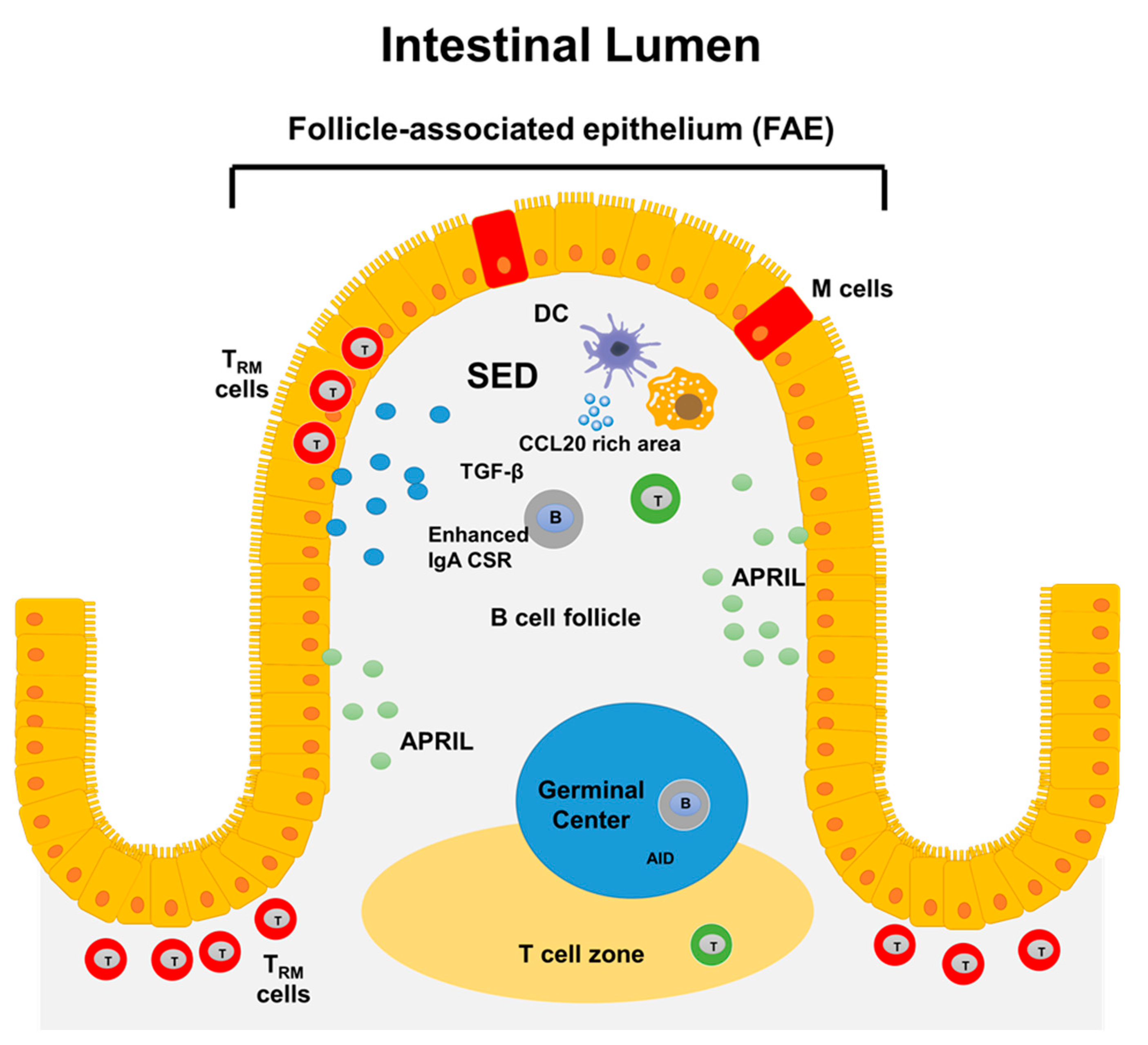
3.1. Gut-Associated Lymphoid Tissues (GALT): Structure and Function
3.2. B Cells in the Gut
3.2.1. Intestinal IgA functions and Lamina Propria PCs
3.2.2. Tissue-Based Memory B Cells
3.2.3. Evidence for PC Survival Niches in the Intestine
3.3. Intestinal Resident T Cells
3.3.1. CD4+ TRM
3.3.2. CD8+ TRM
3.3.3. Regulatory T Cells
4. Respiratory Tract
4.1. Lymphoid Tissues in the Respiratory Track: Organization and Structures
4.1.1. Secretory IgA and Respiratory B Cells
4.1.2. Tissue Resident Memory B Cells in Lungs
4.1.3. B Cell Responses after Mucosal Immunization
4.2. Lung Tissue Resident Memory T (TRM) Cells
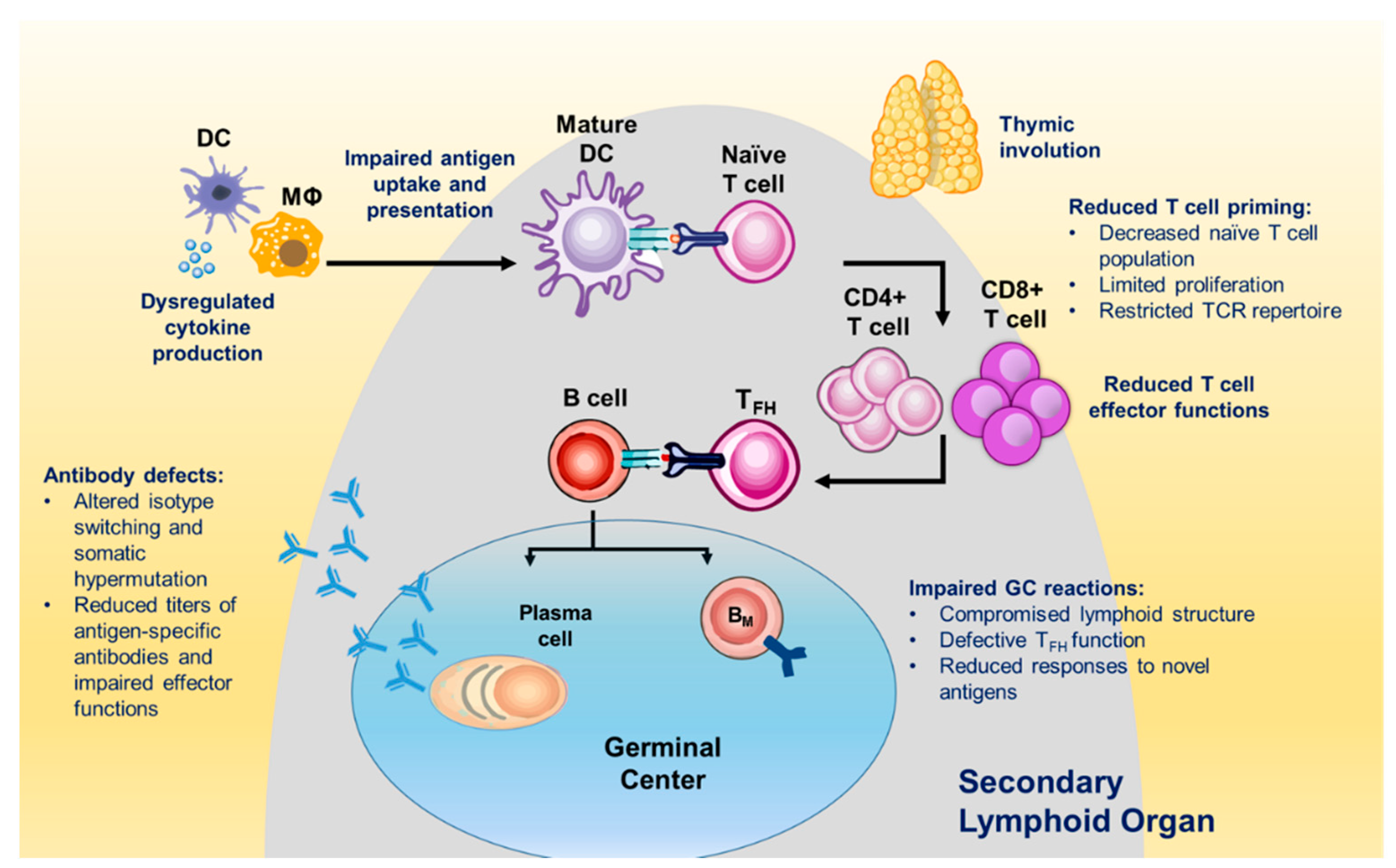
5. Age-Associated Changes in the B- and T-Cell Compartments
5.1. Impact of Aging on B Cells
5.2. Impact of Aging on T Cells
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, I.R. Activation of benign autoimmunity as both tumor and autoimmune disease immunotherapy: A comprehensive review. J Autoimmun. 2014, 54, 112–117. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.A. Skin-resident T cells: The ups and downs of on site immunity. J. Invest. Dermatol. 2010, 130, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Ganusov, V.V.; De Boer, R.J. Do most lymphocytes in humans really reside in the gut? Trends Immunol. 2007, 28, 514–518. [Google Scholar] [CrossRef]
- Lund, F.E.; Garvy, B.A.; Randall, T.D.; Harris, D.P. Regulatory roles for cytokine-producing B cells in infection and autoimmune disease. Curr Dir. Autoimmun. 2005, 8, 25–54. [Google Scholar] [CrossRef]
- Lund, F.E. Cytokine-producing B lymphocytes-key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Shen, P.; Fillatreau, S. Antibody-independent functions of B cells: A focus on cytokines. Nat. Rev. Immunol. 2015, 15, 441–451. [Google Scholar] [CrossRef]
- Lino, A.C.; Dorner, T.; Bar-Or, A.; Fillatreau, S. Cytokine-producing B cells: A translational view on their roles in human and mouse autoimmune diseases. Immunol. Rev. 2016, 269, 130–144. [Google Scholar] [CrossRef]
- Odendahl, M.; Mei, H.; Hoyer, B.F.; Jacobi, A.M.; Hansen, A.; Muehlinghaus, G.; Berek, C.; Hiepe, F.; Manz, R.; Radbruch, A.; et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 2005, 105, 1614–1621. [Google Scholar] [CrossRef]
- Toapanta, F.R.; Simon, J.K.; Barry, E.M.; Pasetti, M.F.; Levine, M.M.; Kotloff, K.L.; Sztein, M.B. Gut-homing conventional plasmablasts and CD27- plasmablasts elicited after a short time exposure to an oral live attenuated Shigella vaccine candidate in humans. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Fink, K. Origin and Function of Circulating Plasmablasts during Acute Viral Infections. Front. Immunol. 2012, 3, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyu, S.Y.; Kobie, J.; Yang, H.; Zand, M.S.; Topham, D.J.; Quataert, S.A.; Sanz, I.; Lee, F.E. Frequencies of human influenza-specific antibody secreting cells or plasmablasts post vaccination from fresh and frozen peripheral blood mononuclear cells. J. Immunol. Methods 2009, 340, 42–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrammert, J.; Onlamoon, N.; Akondy, R.S.; Perng, G.C.; Polsrila, K.; Chandele, A.; Kwissa, M.; Pulendran, B.; Wilson, P.C.; Wittawatmongkol, O.; et al. Rapid and massive virus-specific plasmablast responses during acute dengue virus infection in humans. J. Virol. 2012, 86, 2911–2918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.Q.; Wohlbold, T.J.; Zheng, N.Y.; Huang, M.; Huang, Y.; Neu, K.E.; Lee, J.; Wan, H.; Rojas, K.T.; Kirkpatrick, E.; et al. Influenza Infection in Humans Induces Broadly Cross-Reactive and Protective Neuraminidase-Reactive Antibodies. Cell 2018, 173, 417–429.e410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.Y.; Mays, I.; Garman, L.; Helms, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef] [Green Version]
- Medina, F.; Segundo, C.; Campos-Caro, A.; Gonzalez-Garcia, I.; Brieva, J.A. The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood 2002, 99, 2154–2161. [Google Scholar] [CrossRef] [Green Version]
- Wrammert, J.; Koutsonanos, D.; Li, G.M.; Edupuganti, S.; Sui, J.; Morrissey, M.; McCausland, M.; Skountzou, I.; Hornig, M.; Lipkin, W.I.; et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp Med. 2011, 208, 181–193. [Google Scholar] [CrossRef]
- Turner, J.S.; Zhou, J.Q.; Han, J.; Schmitz, A.J.; Rizk, A.A.; Alsoussi, W.B.; Lei, T.; Amor, M.; McIntire, K.M.; Meade, P.; et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature 2020, 586, 127–132. [Google Scholar] [CrossRef]
- Gonzalez-Garcia, I.; Ocana, E.; Jimenez-Gomez, G.; Campos-Caro, A.; Brieva, J.A. Immunization-induced perturbation of human blood plasma cell pool: Progressive maturation, IL-6 responsiveness, and high PRDI-BF1/BLIMP1 expression are critical distinctions between antigen-specific and nonspecific plasma cells. J. Immunol. 2006, 176, 4042–4050. [Google Scholar] [CrossRef]
- Ellebedy, A.H.; Jackson, K.J.; Kissick, H.T.; Nakaya, H.I.; Davis, C.W.; Roskin, K.M.; McElroy, A.K.; Oshansky, C.M.; Elbein, R.; Thomas, S.; et al. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat. Immunol. 2016, 17, 1226–1234. [Google Scholar] [CrossRef]
- Roldan, E.; Garcia-Pardo, A.; Brieva, J.A. VLA-4-fibronectin interaction is required for the terminal differentiation of human bone marrow cells capable of spontaneous and high rate immunoglobulin secretion. J. Exp. Med. 1992, 175, 1739–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnaure, G.; Gervais-St-Amour, C.; Neron, S. Bone Marrow Mesenchymal Stem Cells Enhance the Differentiation of Human Switched Memory B Lymphocytes into Plasma Cells in Serum-Free Medium. J. Immunol. Res. 2016, 2016, 7801781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabera, S.; Perez-Simon, J.A.; Diez-Campelo, M.; Sanchez-Abarca, L.I.; Blanco, B.; Lopez, A.; Benito, A.; Ocio, E.; Sanchez-Guijo, F.M.; Canizo, C.; et al. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B-lymphocytes. Haematologica 2008, 93, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Berek, C. The establishment of the plasma cell survival niche in the bone marrow. Immunol. Rev. 2013, 251, 177–188. [Google Scholar] [CrossRef]
- Nakayama, T.; Hieshima, K.; Izawa, D.; Tatsumi, Y.; Kanamaru, A.; Yoshie, O. Cutting edge: Profile of chemokine receptor expression on human plasma cells accounts for their efficient recruitment to target tissues. J. Immunol. 2003, 170, 1136–1140. [Google Scholar] [CrossRef] [Green Version]
- Ellyard, J.I.; Avery, D.T.; Mackay, C.R.; Tangye, S.G. Contribution of stromal cells to the migration, function and retention of plasma cells in human spleen: Potential roles of CXCL12, IL-6 and CD54. Eur. J. Immunol. 2005, 35, 699–708. [Google Scholar] [CrossRef]
- Minges Wols, H.A.; Ippolito, J.A.; Yu, Z.; Palmer, J.L.; White, F.A.; Le, P.T.; Witte, P.L. The effects of microenvironment and internal programming on plasma cell survival. Int. Immunol. 2007, 19, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.Y.; Rivas-Caicedo, A.; Renevey, F.; Cannelle, H.; Peranzoni, E.; Scarpellino, L.; Hardie, D.L.; Pommier, A.; Schaeuble, K.; Favre, S.; et al. Identification of a new subset of lymph node stromal cells involved in regulating plasma cell homeostasis. Proc. Natl. Acad. Sci. USA 2018, 115, E6826–E6835. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S.; Aubert, R.D.; Glidewell, J.; Ahmed, R. Tracking human antigen-specific memory B cells: A sensitive and generalized ELISPOT system. J. Immunol. Methods 2004, 286, 111–122. [Google Scholar] [CrossRef]
- Seifert, M.; Przekopowitz, M.; Taudien, S.; Lollies, A.; Ronge, V.; Drees, B.; Lindemann, M.; Hillen, U.; Engler, H.; Singer, B.B.; et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proc. Natl. Acad. Sci. USA 2015, 112, E546–E555. [Google Scholar] [CrossRef] [Green Version]
- Lau, D.; Lan, L.Y.; Andrews, S.F.; Henry, C.; Rojas, K.T.; Neu, K.E.; Huang, M.; Huang, Y.; DeKosky, B.; Palm, A.E.; et al. Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pape, K.A.; Taylor, J.J.; Maul, R.W.; Gearhart, P.J.; Jenkins, M.K. Different B Cell Populations Mediate Early and Late Memory During an Endogenous Immune Response. Science 2011, 331, 1203–1207. [Google Scholar] [CrossRef] [Green Version]
- McHeyzer-Williams, L.J.; Milpied, P.J.; Okitsu, S.L.; McHeyzer-Williams, M.G. Class-switched memory B cells remodel BCRs within secondary germinal centers. Nat. Immunol. 2015, 16, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Dogan, I.; Bertocci, B.; Vilmont, V.; Delbos, F.; Megret, J.; Storck, S.; Reynaud, C.A.; Weill, J.C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009, 10, 1292–1299. [Google Scholar] [CrossRef]
- Jung, J.; Choe, J.; Li, L.; Choi, Y.S. Regulation of CD27 expression in the course of germinal center B cell differentiation: The pivotal role of IL-10. Eur. J. Immunol. 2000, 30, 2437–2443. [Google Scholar] [CrossRef]
- Przekopowitz, M.; Kuppers, R.; Weniger, M.A. A large fraction of human tonsillar B cells expressing CD27 are germinal center B cells. Immunol. Cell Biol. 2015, 93, 429–430. [Google Scholar] [CrossRef]
- Agematsu, K.; Hokibara, S.; Nagumo, H.; Komiyama, A. CD27: A memory B-cell marker. Immunol. Today 2000, 21, 204–206. [Google Scholar] [CrossRef]
- Klein, U.; Rajewsky, K.; Kuppers, R. Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J. Exp. Med. 1998, 188, 1679–1689. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Avery, D.T.; Deenick, E.K.; Hodgkin, P.D. Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. J. Immunol. 2003, 170, 686–694. [Google Scholar] [CrossRef]
- Giesecke, C.; Frolich, D.; Reiter, K.; Mei, H.E.; Wirries, I.; Kuhly, R.; Killig, M.; Glatzer, T.; Stolzel, K.; Perka, C.; et al. Tissue distribution and dependence of responsiveness of human antigen-specific memory B cells. J. Immunol. 2014, 192, 3091–3100. [Google Scholar] [CrossRef] [Green Version]
- Kardava, L.; Moir, S.; Shah, N.; Wang, W.; Wilson, R.; Buckner, C.M.; Santich, B.H.; Kim, L.J.; Spurlin, E.E.; Nelson, A.K.; et al. Abnormal B cell memory subsets dominate HIV-specific responses in infected individuals. J. Clin. Investig. 2014, 124, 3252–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adlowitz, D.G.; Barnard, J.; Biear, J.N.; Cistrone, C.; Owen, T.; Wang, W.; Palanichamy, A.; Ezealah, E.; Campbell, D.; Wei, C.; et al. Expansion of Activated Peripheral Blood Memory B Cells in Rheumatoid Arthritis, Impact of B Cell Depletion Therapy, and Biomarkers of Response. PLoS ONE 2015, 10, e0128269. [Google Scholar] [CrossRef] [Green Version]
- Menard, L.C.; Habte, S.; Gonsiorek, W.; Lee, D.; Banas, D.; Holloway, D.A.; Manjarrez-Orduno, N.; Cunningham, M.; Stetsko, D.; Casano, F.; et al. B cells from African American lupus patients exhibit an activated phenotype. JCI Insight 2016, 1, e87310. [Google Scholar] [CrossRef] [Green Version]
- Glauzy, S.; Boccitto, M.; Bannock, J.M.; Delmotte, F.R.; Saadoun, D.; Cacoub, P.; Ice, J.A.; Sivils, K.L.; James, J.A.; Wolin, S.L.; et al. Accumulation of Antigen-Driven Lymphoproliferations in Complement Receptor 2/CD21(-/low) B Cells From Patients With Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Moir, S.; Ho, J.; Malaspina, A.; Wang, W.; DiPoto, A.C.; O’Shea, M.A.; Roby, G.; Kottilil, S.; Arthos, J.; Proschan, M.A.; et al. Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J. Exp. Med. 2008, 205, 1797–1805. [Google Scholar] [CrossRef]
- Weiss, G.E.; Crompton, P.D.; Li, S.; Walsh, L.A.; Moir, S.; Traore, B.; Kayentao, K.; Ongoiba, A.; Doumbo, O.K.; Pierce, S.K. Atypical memory B cells are greatly expanded in individuals living in a malaria-endemic area. J. Immunol. 2009, 183, 2176–2182. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Anolik, J.; Cappione, A.; Zheng, B.; Pugh-Bernard, A.; Brooks, J.; Lee, E.H.; Milner, E.C.; Sanz, I. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J. Immunol. 2007, 178, 6624–6633. [Google Scholar] [CrossRef] [Green Version]
- Gourley, T.S.; Wherry, E.J.; Masopust, D.; Ahmed, R. Generation and maintenance of immunological memory. Semin. Immunol. 2004, 16, 323–333. [Google Scholar] [CrossRef]
- Kaech, S.M.; Wherry, E.J. Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 2007, 27, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Willinger, T.; Freeman, T.; Hasegawa, H.; McMichael, A.J.; Callan, M.F. Molecular signatures distinguish human central memory from effector memory CD8 T cell subsets. J. Immunol. 2005, 175, 5895–5903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Teichgraber, V.; Becker, T.C.; Masopust, D.; Kaech, S.M.; Antia, R.; von Andrian, U.H.; Ahmed, R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat. Immunol. 2003, 4, 225–234. [Google Scholar] [CrossRef]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Durek, P.; Nordstrom, K.; Gasparoni, G.; Salhab, A.; Kressler, C.; de Almeida, M.; Bassler, K.; Ulas, T.; Schmidt, F.; Xiong, J.; et al. Epigenomic Profiling of Human CD4(+) T Cells Supports a Linear Differentiation Model and Highlights Molecular Regulators of Memory Development. Immunity 2016, 45, 1148–1161. [Google Scholar] [CrossRef] [Green Version]
- Abdelsamed, H.A.; Moustaki, A.; Fan, Y.; Dogra, P.; Ghoneim, H.E.; Zebley, C.C.; Triplett, B.M.; Sekaly, R.P.; Youngblood, B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med. 2017, 214, 1593–1606. [Google Scholar] [CrossRef]
- Moskowitz, D.M.; Zhang, D.W.; Hu, B.; Le Saux, S.; Yanes, R.E.; Ye, Z.; Buenrostro, J.D.; Weyand, C.M.; Greenleaf, W.J.; Goronzy, J.J. Epigenomics of human CD8 T cell differentiation and aging. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef]
- Fresnay, S.; McArthur, M.A.; Magder, L.; Darton, T.C.; Jones, C.; Waddington, C.S.; Blohmke, C.J.; Angus, B.; Levine, M.M.; Pollard, A.J.; et al. Salmonella Typhi-specific multifunctional CD8+ T cells play a dominant role in protection from typhoid fever in humans. J. Transl. Med. 2016, 14, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salerno-Goncalves, R.; Pasetti, M.F.; Sztein, M.B. Characterization of CD8(+) effector T cell responses in volunteers immunized with Salmonella enterica serovar Typhi strain Ty21a typhoid vaccine. J. Immunol. 2002, 169, 2196–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salerno-Goncalves, R.; Wahid, R.; Sztein, M.B. Immunization of volunteers with Salmonella enterica serovar Typhi strain Ty21a elicits the oligoclonal expansion of CD8+ T cells with predominant Vbeta repertoires. Infect. Immun. 2005, 73, 3521–3530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sztein, M.B. Cell-mediated immunity and antibody responses elicited by attenuated Salmonella enterica Serovar Typhi strains used as live oral vaccines in humans. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2007, 45 (Suppl. S1), S15–S19. [Google Scholar] [CrossRef] [Green Version]
- Sztein, M.B.; Salerno-Goncalves, R.; McArthur, M.A. Complex adaptive immunity to enteric fevers in humans: Lessons learned and the path forward. Front. Immunol. 2014, 5, 516. [Google Scholar] [CrossRef]
- Sztein, M.B.; Tanner, M.K.; Polotsky, Y.; Orenstein, J.M.; Levine, M.M. Cytotoxic T lymphocytes after oral immunization with attenuated vaccine strains of Salmonella typhi in humans. J. Immunol. 1995, 155, 3987–3993. [Google Scholar]
- Wahid, R.; Fresnay, S.; Levine, M.M.; Sztein, M.B. Cross-reactive multifunctional CD4+ T cell responses against Salmonella enterica serovars Typhi, Paratyphi A and Paratyphi B in humans following immunization with live oral typhoid vaccine Ty21a. Clin. Immunol. 2016, 173, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Wahid, R.; Salerno-Goncalves, R.; Tacket, C.O.; Levine, M.M.; Sztein, M.B. Generation of specific effector and memory T cells with gut- and secondary lymphoid tissue- homing potential by oral attenuated CVD 909 typhoid vaccine in humans. Mucosal Immunol 2008, 1, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Salerno-Goncalves, R.; Wahid, R.; Sztein, M.B. Ex Vivo kinetics of early and long-term multifunctional human leukocyte antigen E-specific CD8+ cells in volunteers immunized with the Ty21a typhoid vaccine. Clin. Vaccine Immunol. CVI 2010, 17, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Wahid, R.; Fresnay, S.; Levine, M.M.; Sztein, M.B. Immunization with Ty21a live oral typhoid vaccine elicits crossreactive multifunctional CD8+ T-cell responses against Salmonella enterica serovar Typhi, S. Paratyphi A, and S. Paratyphi B in humans. Mucosal Immunol. 2015, 8, 1349–1359. [Google Scholar] [CrossRef] [Green Version]
- Cannons, J.L.; Lu, K.T.; Schwartzberg, P.L. T follicular helper cell diversity and plasticity. Trends Immunol. 2013, 34, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Niro, R.; Lee, S.J.; Vander Heiden, J.A.; Elsner, R.A.; Trivedi, N.; Bannock, J.M.; Gupta, N.T.; Kleinstein, S.H.; Vigneault, F.; Gilbert, T.J.; et al. Salmonella Infection Drives Promiscuous B Cell Activation Followed by Extrafollicular Affinity Maturation. Immunity 2015, 43, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, E.B.; Caporale, L.H.; Thorbecke, G.J. Effect of thymus cell injections on germinal center formation in lymphoid tissues of nude (thymusless) mice. Cell. Immunol. 1974, 13, 416–430. [Google Scholar] [CrossRef]
- Mitchison, N.A. T-cell-B-cell cooperation. Nat. Rev. Immunol. 2004, 4, 308–312. [Google Scholar] [CrossRef]
- Wong, M.T.; Chen, J.; Narayanan, S.; Lin, W.; Anicete, R.; Kiaang, H.T.; De Lafaille, M.A.; Poidinger, M.; Newell, E.W. Mapping the Diversity of Follicular Helper T Cells in Human Blood and Tonsils Using High-Dimensional Mass Cytometry Analysis. Cell Rep. 2015, 11, 1822–1833. [Google Scholar] [CrossRef] [Green Version]
- Fagarasan, S.; Kawamoto, S.; Kanagawa, O.; Suzuki, K. Adaptive immune regulation in the gut: T cell-dependent and T cell-independent IgA synthesis. Annu. Rev. Immunol. 2010, 28, 243–273. [Google Scholar] [CrossRef]
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 2016, 352, aaf4822. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.L. Uptake and transport of intestinal macromolecules and microorganisms by M cells in Peyer’s patches—A personal and historical perspective. Semin. Immunol. 1999, 11, 157–163. [Google Scholar] [CrossRef]
- Neutra, M.R.; Pringault, E.; Kraehenbuhl, J.P. Antigen sampling across epithelial barriers and induction of mucosal immune responses. Annu. Rev. Immunol. 1996, 14, 275–300. [Google Scholar] [CrossRef]
- Rochereau, N.; Drocourt, D.; Perouzel, E.; Pavot, V.; Redelinghuys, P.; Brown, G.D.; Tiraby, G.; Roblin, X.; Verrier, B.; Genin, C.; et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013, 11, e1001658. [Google Scholar] [CrossRef]
- Reboldi, A.; Cyster, J.G. Peyer’s patches: Organizing B-cell responses at the intestinal frontier. Immunol. Rev. 2016, 271, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Gebert, A.; Steinmetz, I.; Fassbender, S.; Wendlandt, K.H. Antigen transport into Peyer’s patches: Increased uptake by constant numbers of M cells. Am. J. Pathol. 2004, 164, 65–72. [Google Scholar] [CrossRef]
- Heel, K.A.; McCauley, R.D.; Papadimitriou, J.M.; Hall, J.C. Review: Peyer’s patches. J. Gastroenterol. Hepatol. 1997, 12, 122–136. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, M.; Hase, K.; Takahashi, D.; Kitamura, H.; Knoop, K.A.; Williams, I.R.; Ohno, H. CCR6hiCD11c(int) B cells promote M-cell differentiation in Peyer’s patch. Int. Immunol. 2011, 23, 261–269. [Google Scholar] [CrossRef]
- Varona, R.; Villares, R.; Carramolino, L.; Goya, I.; Zaballos, A.; Gutierrez, J.; Torres, M.; Martinez, A.C.; Marquez, G. CCR6-deficient mice have impaired leukocyte homeostasis and altered contact hypersensitivity and delayed-type hypersensitivity responses. J. Clin. Invest. 2001, 107, R37–R45. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.N.; Prosser, D.M.; Forster, R.; Zhang, J.; Kuklin, N.A.; Abbondanzo, S.J.; Niu, X.D.; Chen, S.C.; Manfra, D.J.; Wiekowski, M.T.; et al. CCR6 mediates dendritic cell localization, lymphocyte homeostasis, and immune responses in mucosal tissue. Immunity 2000, 12, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Travis, M.A.; Reizis, B.; Melton, A.C.; Masteller, E.; Tang, Q.; Proctor, J.M.; Wang, Y.; Bernstein, X.; Huang, X.; Reichardt, L.F.; et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 2007, 449, 361–365. [Google Scholar] [CrossRef]
- Borsutzky, S.; Cazac, B.B.; Roes, J.; Guzman, C.A. TGF-beta receptor signaling is critical for mucosal IgA responses. J. Immunol. 2004, 173, 3305–3309. [Google Scholar] [CrossRef] [Green Version]
- Cazac, B.B.; Roes, J. TGF-beta receptor controls B cell responsiveness and induction of IgA in vivo. Immunity 2000, 13, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Tangye, S.G.; Tarlinton, D.M. Memory B cells: Effectors of long-lived immune responses. Eur. J. Immunol. 2009, 39, 2065–2075. [Google Scholar] [CrossRef]
- Brandtzaeg, P. Update on mucosal immunoglobulin A in gastrointestinal disease. Curr. Opin. Gastroenterol. 2010, 26, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lycke, N.; Erlandsson, L.; Ekman, L.; Schon, K.; Leanderson, T. Lack of J chain inhibits the transport of gut IgA and abrogates the development of intestinal antitoxic protection. J. Immunol. 1999, 163, 913–919. [Google Scholar] [PubMed]
- Forbes, S.J.; Eschmann, M.; Mantis, N.J. Inhibition of Salmonella enterica serovar typhimurium motility and entry into epithelial cells by a protective antilipopolysaccharide monoclonal immunoglobulin A antibody. Infect. Immun. 2008, 76, 4137–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.J.; Bumpus, T.; McCarthy, E.A.; Corthesy, B.; Mantis, N.J. Transient suppression of Shigella flexneri type 3 secretion by a protective O-antigen-specific monoclonal IgA. mBio 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadaoui, K.A.; Corthesy, B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer’s patches with restriction to mucosal compartment. J. Immunol. 2007, 179, 7751–7757. [Google Scholar] [CrossRef] [Green Version]
- Bakema, J.E.; van Egmond, M. The human immunoglobulin A Fc receptor FcalphaRI: A multifaceted regulator of mucosal immunity. Mucosal Immunol. 2011, 4, 612–624. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.K.; Blanchard, T.G.; Levine, A.D.; Emancipator, S.N.; Lamm, M.E. A mucosal IgA-mediated excretory immune system in vivo. J. Immunol. 2001, 166, 3688–3692. [Google Scholar] [CrossRef] [Green Version]
- Farstad, I.N.; Carlsen, H.; Morton, H.C.; Brandtzaeg, P. Immunoglobulin A cell distribution in the human small intestine: Phenotypic and functional characteristics. Immunology 2000, 101, 354–363. [Google Scholar] [CrossRef]
- Brandtzaeg, P.; Farstad, I.N.; Johansen, F.E.; Morton, H.C.; Norderhaug, I.N.; Yamanaka, T. The B-cell system of human mucosae and exocrine glands. Immunol. Rev. 1999, 171, 45–87. [Google Scholar] [CrossRef]
- Nair, N.; Newell, E.W.; Vollmers, C.; Quake, S.R.; Morton, J.M.; Davis, M.M.; He, X.S.; Greenberg, H.B. High-dimensional immune profiling of total and rotavirus VP6-specific intestinal and circulating B cells by mass cytometry. Mucosal Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P. Function of mucosa-associated lymphoid tissue in antibody formation. Immunol. Invest. 2010, 39, 303–355. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Tiacci, E.; Pucciarini, A.; Bigerna, B.; Kurth, J.; Hatzivassiliou, G.; Droetto, S.; Galletti, B.V.; Gambacorta, M.; Orazi, A.; et al. Expression of the IRTA1 receptor identifies intraepithelial and subepithelial marginal zone B cells of the mucosa-associated lymphoid tissue (MALT). Blood 2003, 102, 3684–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrhardt, G.R.; Davis, R.S.; Hsu, J.T.; Leu, C.M.; Ehrhardt, A.; Cooper, M.D. The inhibitory potential of Fc receptor homolog 4 on memory B cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13489–13494. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, G.R.; Hsu, J.T.; Gartland, L.; Leu, C.M.; Zhang, S.; Davis, R.S.; Cooper, M.D. Expression of the immunoregulatory molecule FcRH4 defines a distinctive tissue-based population of memory B cells. J. Exp. Med. 2005, 202, 783–791. [Google Scholar] [CrossRef]
- Sohn, H.W.; Krueger, P.D.; Davis, R.S.; Pierce, S.K. FcRL4 acts as an adaptive to innate molecular switch dampening BCR signaling and enhancing TLR signaling. Blood 2011, 118, 6332–6341. [Google Scholar] [CrossRef] [Green Version]
- Mesin, L.; Di Niro, R.; Thompson, K.M.; Lundin, K.E.; Sollid, L.M. Long-lived plasma cells from human small intestine biopsies secrete immunoglobulins for many weeks in vitro. J. Immunol. 2011, 187, 2867–2874. [Google Scholar] [CrossRef] [Green Version]
- Barone, F.; Patel, P.; Sanderson, J.D.; Spencer, J. Gut-associated lymphoid tissue contains the molecular machinery to support T-cell-dependent and T-cell-independent class switch recombination. Mucosal Immunol. 2009, 2, 495–503. [Google Scholar] [CrossRef]
- Ng, E.K.; Panesar, N.; Longo, W.E.; Shapiro, M.J.; Kaminski, D.L.; Tolman, K.C.; Mazuski, J.E. Human intestinal epithelial and smooth muscle cells are potent producers of IL-6. Mediators Inflamm. 2003, 12, 3–8. [Google Scholar] [CrossRef]
- Dotan, I.; Werner, L.; Vigodman, S.; Weiss, S.; Brazowski, E.; Maharshak, N.; Chen, O.; Tulchinsky, H.; Halpern, Z.; Guzner-Gur, H. CXCL12 is a constitutive and inflammatory chemokine in the intestinal immune system. Inflamm. Bowel Dis. 2010, 16, 583–592. [Google Scholar] [CrossRef]
- Thome, J.J.; Farber, D.L. Emerging concepts in tissue-resident T cells: Lessons from humans. Trends Immunol. 2015, 36, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathaliyawala, T.; Kubota, M.; Yudanin, N.; Turner, D.; Camp, P.; Thome, J.J.; Bickham, K.L.; Lerner, H.; Goldstein, M.; Sykes, M.; et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 2013, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Gehad, A.; Yang, C.; Scott, L.L.; Teague, J.E.; Schlapbach, C.; Elco, C.P.; Huang, V.; Matos, T.R.; Kupper, T.S.; et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 2015, 7, 279ra239. [Google Scholar] [CrossRef] [Green Version]
- Hombrink, P.; Helbig, C.; Backer, R.A.; Piet, B.; Oja, A.E.; Stark, R.; Brasser, G.; Jongejan, A.; Jonkers, R.E.; Nota, B.; et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat. Immunol. 2016, 17, 1467–1478. [Google Scholar] [CrossRef]
- Oja, A.E.; Piet, B.; Helbig, C.; Stark, R.; van der Zwan, D.; Blaauwgeers, H.; Remmerswaal, E.B.M.; Amsen, D.; Jonkers, R.E.; Moerland, P.D.; et al. Trigger-happy resident memory CD4(+) T cells inhabit the human lungs. Mucosal Immunol. 2018, 11, 654–667. [Google Scholar] [CrossRef]
- Oja, A.E.; Piet, B.; van der Zwan, D.; Blaauwgeers, H.; Mensink, M.; de Kivit, S.; Borst, J.; Nolte, M.A.; van Lier, R.A.W.; Stark, R.; et al. Functional Heterogeneity of CD4(+) Tumor-Infiltrating Lymphocytes With a Resident Memory Phenotype in NSCLC. Front. Immunol. 2018, 9, 2654. [Google Scholar] [CrossRef] [Green Version]
- Park, C.O.; Fu, X.; Jiang, X.; Pan, Y.; Teague, J.E.; Collins, N.; Tian, T.; O’Malley, J.T.; Emerson, R.O.; Kim, J.H.; et al. Staged development of long-lived T-cell receptor alphabeta TH17 resident memory T-cell population to Candida albicans after skin infection. J. Allergy Clin. Immunol. 2018, 142, 647–662. [Google Scholar] [CrossRef] [Green Version]
- Moylan, D.C.; Goepfert, P.A.; Kempf, M.C.; Saag, M.S.; Richter, H.E.; Mestecky, J.; Sabbaj, S. Diminished CD103 (alphaEbeta7) Expression on Resident T Cells from the Female Genital Tract of HIV-Positive Women. Pathog. Immun. 2016, 1, 371–387. [Google Scholar] [CrossRef] [Green Version]
- Rochman, Y.; Spolski, R.; Leonard, W.J. New insights into the regulation of T cells by gamma(c) family cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Kalia, V.; Sarkar, S.; Subramaniam, S.; Haining, W.N.; Smith, K.A.; Ahmed, R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 2010, 32, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipkin, M.E.; Sacks, J.A.; Cruz-Guilloty, F.; Lichtenheld, M.G.; Bevan, M.J.; Rao, A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 2010, 32, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Dhume, K.; McKinstry, K.K. Early programming and late-acting checkpoints governing the development of CD4 T-cell memory. Immunology 2018, 155, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strutt, T.M.; Dhume, K.; Finn, C.M.; Hwang, J.H.; Castonguay, C.; Swain, S.L.; McKinstry, K.K. IL-15 supports the generation of protective lung-resident memory CD4 T cells. Mucosal Immunol. 2018, 11, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Raeber, M.E.; Zurbuchen, Y.; Impellizzieri, D.; Boyman, O. The role of cytokines in T-cell memory in health and disease. Immunol. Rev. 2018, 283, 176–193. [Google Scholar] [CrossRef]
- Yeon, S.M.; Halim, L.; Chandele, A.; Perry, C.J.; Kim, S.H.; Kim, S.U.; Byun, Y.; Yuk, S.H.; Kaech, S.M.; Jung, Y.W. IL-7 plays a critical role for the homeostasis of allergen-specific memory CD4 T cells in the lung and airways. Sci. Rep. 2017, 7, 11155. [Google Scholar] [CrossRef] [Green Version]
- Collins, N.; Jiang, X.; Zaid, A.; Macleod, B.L.; Li, J.; Park, C.O.; Haque, A.; Bedoui, S.; Heath, W.R.; Mueller, S.N.; et al. Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun. 2016, 7, 11514. [Google Scholar] [CrossRef]
- Iijima, N.; Iwasaki, A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 2014, 346, 93–98. [Google Scholar] [CrossRef] [Green Version]
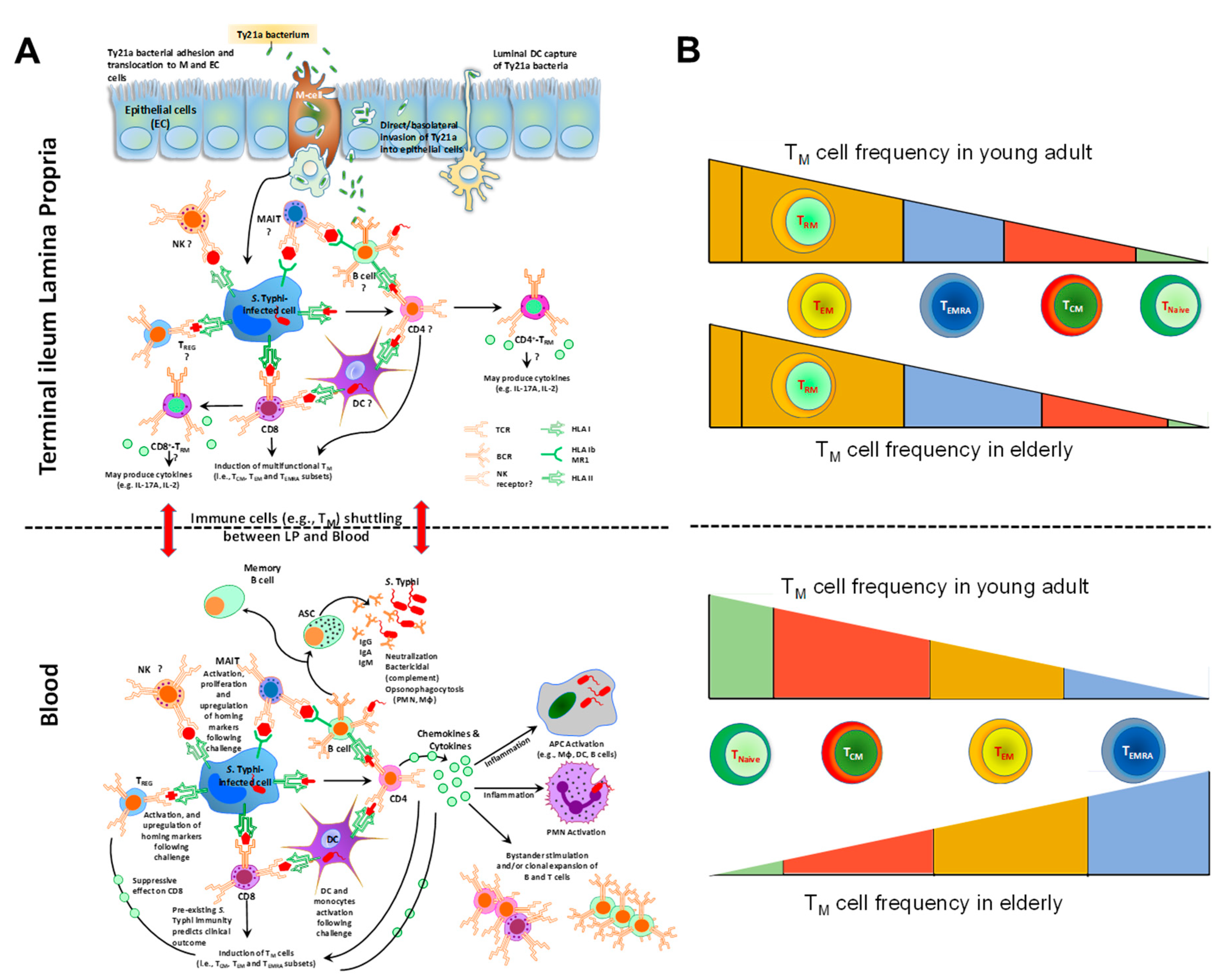
- Booth, J.S.; Goldberg, E.; Barnes, R.S.; Greenwald, B.D.; Sztein, M.B. Oral typhoid vaccine Ty21a elicits antigen-specific resident memory CD4(+) T cells in the human terminal ileum lamina propria and epithelial compartments. J. Transl. Med. 2020, 18, 102. [Google Scholar] [CrossRef]
- Toapanta, F.R.; Booth, J.S.; Sztein, M.B. Chapter 29-Induction of Local and Systemic Immunity by Salmonella Typhi in Humans. In Mucosal Vaccines, 2nd ed.; Kiyono, H., Pascual, D.W., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 501–513. [Google Scholar] [CrossRef]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 2012, 483, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Bevan, M.J.; Fink, P.J. Local Inflammatory Cues Regulate Differentiation and Persistence of CD8(+) Tissue-Resident Memory T Cells. Cell Rep. 2017, 19, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Bank, I.; Book, M.; Ware, R. Functional role of VLA-1 (CD49A) in adhesion, cation-dependent spreading, and activation of cultured human T lymphocytes. Cell. Immunol. 1994, 156, 424–437. [Google Scholar] [CrossRef]
- Roberts, A.I.; Brolin, R.E.; Ebert, E.C. Integrin alpha1beta1 (VLA-1) mediates adhesion of activated intraepithelial lymphocytes to collagen. Immunology 1999, 97, 679–685. [Google Scholar] [CrossRef]
- Ben-Horin, S.; Bank, I. The role of very late antigen-1 in immune-mediated inflammation. Clin. Immunol. 2004, 113, 119–129. [Google Scholar] [CrossRef]
- Budd, R.C.; Cerottini, J.C.; MacDonald, H.R. Phenotypic identification of memory cytolytic T lymphocytes in a subset of Lyt-2+ cells. J. Immunol. 1987, 138, 1009–1013. [Google Scholar]
- Richter, M.; Ray, S.J.; Chapman, T.J.; Austin, S.J.; Rebhahn, J.; Mosmann, T.R.; Gardner, H.; Kotelianski, V.; de Fougerolles, A.R.; Topham, D.J. Collagen distribution and expression of collagen-binding alpha1beta1 (VLA-1) and alpha2beta1 (VLA-2) integrins on CD4 and CD8 T cells during influenza infection. J. Immunol. 2007, 178, 4506–4516. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Koelle, D.M.; Cao, J.; Vazquez, J.; Huang, M.L.; Hladik, F.; Wald, A.; Corey, L. Virus-specific CD8+ T cells accumulate near sensory nerve endings in genital skin during subclinical HSV-2 reactivation. J. Exp. Med. 2007, 204, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Peng, T.; Johnston, C.; Phasouk, K.; Kask, A.S.; Klock, A.; Jin, L.; Diem, K.; Koelle, D.M.; Wald, A.; et al. Immune surveillance by CD8alphaalpha+ skin-resident T cells in human herpes virus infection. Nature 2013, 497, 494–497. [Google Scholar] [CrossRef] [Green Version]
- Purwar, R.; Campbell, J.; Murphy, G.; Richards, W.G.; Clark, R.A.; Kupper, T.S. Resident memory T cells (T(RM)) are abundant in human lung: Diversity, function, and antigen specificity. PLoS ONE 2011, 6, e16245. [Google Scholar] [CrossRef] [Green Version]
- Turner, D.L.; Bickham, K.L.; Thome, J.J.; Kim, C.Y.; D’Ovidio, F.; Wherry, E.J.; Farber, D.L. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 2014, 7, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2(high) tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef] [PubMed]
- Woon, H.G.; Braun, A.; Li, J.; Smith, C.; Edwards, J.; Sierro, F.; Feng, C.G.; Khanna, R.; Elliot, M.; Bell, A.; et al. Compartmentalization of Total and Virus-Specific Tissue-Resident Memory CD8+ T Cells in Human Lymphoid Organs. PLoS Pathog. 2016, 12, e1005799. [Google Scholar] [CrossRef]
- Booth, J.S.; Patil, S.A.; Goldberg, E.; Barnes, R.S.; Greenwald, B.D.; Sztein, M.B. Attenuated Oral Typhoid Vaccine Ty21a Elicits Lamina Propria and Intra-Epithelial Lymphocyte Tissue-Resident Effector Memory CD8 T Responses in the Human Terminal Ileum. Front. Immunol. 2019, 10, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, L.R.; Tsavaler, L.; Rivas, A.; Engleman, E.G. V7 (CD101) ligation inhibits TCR/CD3-induced IL-2 production by blocking Ca2+ flux and nuclear factor of activated T cell nuclear translocation. J. Immunol. 1998, 161, 209–217. [Google Scholar] [PubMed]
- Bergsbaken, T.; Bevan, M.J. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat. Immunol. 2015, 16, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Cheuk, S.; Schlums, H.; Gallais Serezal, I.; Martini, E.; Chiang, S.C.; Marquardt, N.; Gibbs, A.; Detlofsson, E.; Introini, A.; Forkel, M.; et al. CD49a Expression Defines Tissue-Resident CD8(+) T Cells Poised for Cytotoxic Function in Human Skin. Immunity 2017, 46, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef] [Green Version]
- Churov, A.V.; Mamashov, K.Y.; Novitskaia, A.V. Homeostasis and the functional roles of CD4(+) Treg cells in aging. Immunol. Lett. 2020, 226, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Whibley, N.; Tucci, A.; Powrie, F. Regulatory T cell adaptation in the intestine and skin. Nat. Immunol. 2019, 20, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Lui, P.P.; Cho, I.; Ali, N. Tissue regulatory T cells. Immunology 2020, 161, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Panduro, M.; Benoist, C.; Mathis, D. Tissue Tregs. Annu. Rev. Immunol. 2016, 34, 609–633. [Google Scholar] [CrossRef] [Green Version]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A special population of regulatory T cells potentiates muscle repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Bienenstock, J.; McDermott, M.R. Bronchus- and nasal-associated lymphoid tissues. Immunol. Rev. 2005, 206, 22–31. [Google Scholar] [CrossRef]
- Ogasawara, N.; Kojima, T.; Go, M.; Takano, K.; Kamekura, R.; Ohkuni, T.; Koizumi, J.; Masaki, T.; Fuchimoto, J.; Obata, K.; et al. Epithelial barrier and antigen uptake in lymphoepithelium of human adenoids. Acta Otolaryngol. 2011, 131, 116–123. [Google Scholar] [CrossRef]
- Verbrugghe, P.; Kujala, P.; Waelput, W.; Peters, P.J.; Cuvelier, C.A. Clusterin in human gut-associated lymphoid tissue, tonsils, and adenoids: Localization to M cells and follicular dendritic cells. Histochem. Cell Biol. 2008, 129, 311–320. [Google Scholar] [CrossRef]
- Kim, D.Y.; Sato, A.; Fukuyama, S.; Sagara, H.; Nagatake, T.; Kong, I.G.; Goda, K.; Nochi, T.; Kunisawa, J.; Sato, S.; et al. The airway antigen sampling system: Respiratory M cells as an alternative gateway for inhaled antigens. J. Immunol. 2011, 186, 4253–4262. [Google Scholar] [CrossRef] [Green Version]
- Hiller, A.S.; Tschernig, T.; Kleemann, W.J.; Pabst, R. Bronchus-associated lymphoid tissue (BALT) and larynx-associated lymphoid tissue (LALT) are found at different frequencies in children, adolescents and adults. Scand. J. Immunol. 1998, 47, 159–162. [Google Scholar] [CrossRef]
- Richmond, I.; Pritchard, G.E.; Ashcroft, T.; Avery, A.; Corris, P.A.; Walters, E.H. Bronchus associated lymphoid tissue (BALT) in human lung: Its distribution in smokers and non-smokers. Thorax 1993, 48, 1130–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delventhal, S.; Brandis, A.; Ostertag, H.; Pabst, R. Low incidence of bronchus-associated lymphoid tissue (BALT) in chronically inflamed human lungs. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1992, 62, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Cameron, L.; Hamid, Q.; Wright, E.; Nakamura, Y.; Christodoulopoulos, P.; Muro, S.; Frenkiel, S.; Lavigne, F.; Durham, S.; Gould, H. Local synthesis of epsilon germline gene transcripts, IL-4, and IL-13 in allergic nasal mucosa after ex vivo allergen exposure. J. Allergy Clin. Immunol. 2000, 106, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gould, H.J.; Sutton, B.J. IgE in allergy and asthma today. Nat. Rev. Immunol. 2008, 8, 205–217. [Google Scholar] [CrossRef]
- Tezuka, H.; Abe, Y.; Iwata, M.; Takeuchi, H.; Ishikawa, H.; Matsushita, M.; Shiohara, T.; Akira, S.; Ohteki, T. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 2007, 448, 929–933. [Google Scholar] [CrossRef]
- Cerutti, A.; Chen, K.; Chorny, A. Immunoglobulin responses at the mucosal interface. Annu. Rev. Immunol. 2011, 29, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Xu, W.; Wilson, M.; He, B.; Miller, N.W.; Bengten, E.; Edholm, E.S.; Santini, P.A.; Rath, P.; Chiu, A.; et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nat. Immunol. 2009, 10, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.Y.; Wilson, K.; Wang, X.; Boston, A.; Kolar, G.; Jackson, S.M.; Liu, Y.J.; Pascual, V.; Capra, J.D.; Wilson, P.C. Human immunoglobulin selection associated with class switch and possible tolerogenic origins for C delta class-switched B cells. J. Clin. Invest. 2004, 113, 1188–1201. [Google Scholar] [CrossRef] [Green Version]
- Kiyono, H.; Fukuyama, S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef]
- Onodera, T.; Takahashi, Y.; Yokoi, Y.; Ato, M.; Kodama, Y.; Hachimura, S.; Kurosaki, T.; Kobayashi, K. Memory B cells in the lung participate in protective humoral immune responses to pulmonary influenza virus reinfection. Proc. Natl. Acad. Sci. USA 2012, 109, 2485–2490. [Google Scholar] [CrossRef] [Green Version]
- Allie, S.R.; Bradley, J.E.; Mudunuru, U.; Schultz, M.D.; Graf, B.A.; Lund, F.E.; Randall, T.D. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat. Immunol. 2019, 20, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Barker, K.A.; Shenoy, A.T.; Stauffer-Smith, N.; Arafa, E.I.; de Ana, C.L.; Barron, A.; Aihara, F.; Martin, I.; Zhong, X.; Kepler, T.B.; et al. Lung resident memory B cells are a common and functionally significant component of lung adaptive immunity. J. Immunol. 2020, 204, 85.8. [Google Scholar]
- Islam, S.; Mohn, K.G.; Krammer, F.; Sanne, M.; Bredholt, G.; Jul-Larsen, A.; Tete, S.M.; Zhou, F.; Brokstad, K.A.; Cox, R.J. Influenza A haemagglutinin specific IgG responses in children and adults after seasonal trivalent live attenuated influenza vaccination. Vaccine 2017, 35, 5666–5673. [Google Scholar] [CrossRef] [Green Version]
- Babu, T.M.; Levine, M.; Fitzgerald, T.; Luke, C.; Sangster, M.Y.; Jin, H.; Topham, D.; Katz, J.; Treanor, J.; Subbarao, K. Live attenuated H7N7 influenza vaccine primes for a vigorous antibody response to inactivated H7N7 influenza vaccine. Vaccine 2014, 32, 6798–6804. [Google Scholar] [CrossRef]
- Talaat, K.R.; Luke, C.J.; Khurana, S.; Manischewitz, J.; King, L.R.; McMahon, B.A.; Karron, R.A.; Lewis, K.D.; Qin, J.; Follmann, D.A.; et al. A live attenuated influenza A(H5N1) vaccine induces long-term immunity in the absence of a primary antibody response. J. Infect. Dis. 2014, 209, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Sobhanie, M.; Matsuoka, Y.; Jegaskanda, S.; Fitzgerald, T.; Mallory, R.; Chen, Z.; Luke, C.; Treanor, J.; Subbarao, K. Evaluation of the Safety and Immunogenicity of a Candidate Pandemic Live Attenuated Influenza Vaccine (pLAIV) Against Influenza A(H7N9). J. Infect. Dis. 2016, 213, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Jegaskanda, S.; Mason, R.D.; Andrews, S.F.; Wheatley, A.K.; Zhang, R.; Reynoso, G.V.; Ambrozak, D.R.; Santos, C.P.; Luke, C.J.; Matsuoka, Y.; et al. Intranasal Live Influenza Vaccine Priming Elicits Localized B Cell Responses in Mediastinal Lymph Nodes. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Zens, K.D.; Chen, J.K.; Farber, D.L. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2016, 1. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.; Mangalam, A.K.; Channappanavar, R.; Fett, C.; Meyerholz, D.K.; Agnihothram, S.; Baric, R.S.; David, C.S.; Perlman, S. Airway Memory CD4(+) T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity 2016, 44, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Walrath, J.R.; Silver, R.F. The alpha4beta1 integrin in localization of Mycobacterium tuberculosis-specific T helper type 1 cells to the human lung. Am. J. Respir. Cell Mol. Biol. 2011, 45, 24–30. [Google Scholar] [CrossRef]
- McKinstry, K.K.; Strutt, T.M.; Bautista, B.; Zhang, W.; Kuang, Y.; Cooper, A.M.; Swain, S.L. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nature Commun. 2014, 5, 5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.J.; Goldrath, A.W. Transcriptional programming of tissue-resident memory CD8(+) T cells. Curr. Opin. Immunol. 2018, 51, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.C.; Choi, D.H.; Choi, Y.W.; Park, S.J.; Namkoong, H.; Park, K.S.; Ahn, S.S.; Surh, C.D.; Yoon, S.W.; Kim, D.J.; et al. Intranasal Introduction of Fc-Fused Interleukin-7 Provides Long-Lasting Prophylaxis against Lethal Influenza Virus Infection. J. Virol. 2015, 90, 2273–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bree, G.J.; Daniels, H.; Schilfgaarde, M.; Jansen, H.M.; Out, T.A.; van Lier, R.A.; Jonkers, R.E. Characterization of CD4+ memory T cell responses directed against common respiratory pathogens in peripheral blood and lung. J. Infect. Dis. 2007, 195, 1718–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinstry, K.K.; Strutt, T.M.; Kuang, Y.; Brown, D.M.; Sell, S.; Dutton, R.W.; Swain, S.L. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J. Clin. Investig. 2012, 122, 2847–2856. [Google Scholar] [CrossRef] [Green Version]
- Teijaro, J.R.; Verhoeven, D.; Page, C.A.; Turner, D.; Farber, D.L. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J. Virol. 2010, 84, 9217–9226. [Google Scholar] [CrossRef] [Green Version]
- Sakai, S.; Kauffman, K.D.; Schenkel, J.M.; McBerry, C.C.; Mayer-Barber, K.D.; Masopust, D.; Barber, D.L. Cutting edge: Control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 2014, 192, 2965–2969. [Google Scholar] [CrossRef] [Green Version]
- United Nations. World Population Prospects 2019. Available online: https://population.un.org/wpp/ (accessed on 25 November 2020).
- Pera, A.; Campos, C.; Lopez, N.; Hassouneh, F.; Alonso, C.; Tarazona, R.; Solana, R. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 2015, 82, 50–55. [Google Scholar] [CrossRef]
- Joseph, F.; Julia, W. Aging, Immunity, and Infection; Humana Press: Totowa, NJ, USA, 2003. [Google Scholar]
- Scallan, E.; Crim, S.M.; Runkle, A.; Henao, O.L.; Mahon, B.E.; Hoekstra, R.M.; Griffin, P.M. Bacterial Enteric Infections Among Older Adults in the United States: Foodborne Diseases Active Surveillance Network, 1996–2012. Foodborne Pathog. Dis. 2015, 12, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Wroe, P.C.; Finkelstein, J.A.; Ray, G.T.; Linder, J.A.; Johnson, K.M.; Rifas-Shiman, S.; Moore, M.R.; Huang, S.S. Aging population and future burden of pneumococcal pneumonia in the United States. J. Infect. Dis. 2012, 205, 1589–1592. [Google Scholar] [CrossRef]
- Gordon, A.; Reingold, A. The Burden of Influenza: A Complex Problem. Curr. Epidemiol. Rep. 2018, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.W.; Bouhassira, D.; Kassianos, G.; Leplege, A.; Schmader, K.E.; Weinke, T. The impact of herpes zoster and post-herpetic neuralgia on quality-of-life. BMC Med. 2010, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.C.; Toapanta, F.R.; Chen, W.; Tennant, S.M. Understanding immunosenescence and its impact on vaccination of older adults. Vaccine 2020, 38, 8264–8272. [Google Scholar] [CrossRef]
- Kaml, M.; Weiskirchner, I.; Keller, M.; Luft, T.; Hoster, E.; Hasford, J.; Young, L.; Bartlett, B.; Neuner, C.; Fischer, K.H.; et al. Booster vaccination in the elderly: Their success depends on the vaccine type applied earlier in life as well as on pre-vaccination antibody titers. Vaccine 2006, 24, 6808–6811. [Google Scholar] [CrossRef]
- Weinberger, B.; Schirmer, M.; Matteucci Gothe, R.; Siebert, U.; Fuchs, D.; Grubeck-Loebenstein, B. Recall responses to tetanus and diphtheria vaccination are frequently insufficient in elderly persons. PLoS ONE 2013, 8, e82967. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. The generation of memory B cells is maintained, but the antibody response is not, in the elderly after repeated influenza immunizations. Vaccine 2016, 34, 2834–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferson, T.; Rivetti, D.; Rivetti, A.; Rudin, M.; Di Pietrantonj, C.; Demicheli, V. Efficacy and effectiveness of influenza vaccines in elderly people: A systematic review. Lancet 2005, 366, 1165–1174. [Google Scholar] [CrossRef]
- Kositanont, U.; Assantachai, P.; Wasi, C.; Puthavathana, P.; Praditsuwan, R. Kinetics of the antibody response to seasonal influenza vaccination among the elderly. Viral Immunol. 2012, 25, 471–476. [Google Scholar] [CrossRef]
- Henry, C.; Zheng, N.Y.; Huang, M.; Cabanov, A.; Rojas, K.T.; Kaur, K.; Andrews, S.F.; Palm, A.E.; Chen, Y.Q.; Li, Y.; et al. Influenza Virus Vaccination Elicits Poorly Adapted B Cell Responses in Elderly Individuals. Cell Host Microb. 2019, 25, 357–366 e356. [Google Scholar] [CrossRef] [Green Version]
- Frasca, D.; Van der Put, E.; Riley, R.L.; Blomberg, B.B. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J. Immunol. 2004, 172, 2155–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, S.; Frasca, D.; Blomberg, B.; Golding, H. AID activity in B cells strongly correlates with polyclonal antibody affinity maturation in-vivo following pandemic 2009-H1N1 vaccination in humans. PLoS Pathog. 2012, 8, e1002920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; O’Neill, P.; Naradikian, M.S.; Scholz, J.L.; Cancro, M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 2011, 118, 1294–1304. [Google Scholar] [CrossRef] [Green Version]
- Jenks, S.A.; Cashman, K.S.; Zumaquero, E.; Marigorta, U.M.; Patel, A.V.; Wang, X.; Tomar, D.; Woodruff, M.C.; Simon, Z.; Bugrovsky, R.; et al. Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 2018, 49, 725–739.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, I.; Wei, C.; Jenks, S.A.; Cashman, K.S.; Tipton, C.; Woodruff, M.C.; Hom, J.; Lee, F.E. Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 2019, 10, 2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Giudice, G.; Goronzy, J.J.; Grubeck-Loebenstein, B.; Lambert, P.H.; Mrkvan, T.; Stoddard, J.J.; Doherty, T.M. Fighting against a protean enemy: Immunosenescence, vaccines, and healthy aging. NPJ Aging Mech. Dis. 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, K.M.; Fox, A.; Harland, K.L.; Russ, B.E.; Li, J.; Nguyen, T.H.O.; Loh, L.; Olshanksy, M.; Naeem, H.; Tsyganov, K.; et al. Age-Related Decline in Primary CD8(+) T Cell Responses Is Associated with the Development of Senescence in Virtual Memory CD8(+) T Cells. Cell Rep. 2018, 23, 3512–3524. [Google Scholar] [CrossRef]
- Naylor, K.; Li, G.; Vallejo, A.N.; Lee, W.W.; Koetz, K.; Bryl, E.; Witkowski, J.; Fulbright, J.; Weyand, C.M.; Goronzy, J.J. The influence of age on T cell generation and TCR diversity. J. Immunol. 2005, 174, 7446–7452. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Weyand, C.M. T cell development and receptor diversity during aging. Curr. Opin. Immunol. 2005, 17, 468–475. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Thome, J.J.; Bickham, K.L.; Ohmura, Y.; Kubota, M.; Matsuoka, N.; Gordon, C.; Granot, T.; Griesemer, A.; Lerner, H.; Kato, T.; et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med. 2016, 22, 72–77. [Google Scholar] [CrossRef]
- Thome, J.J.; Yudanin, N.; Ohmura, Y.; Kubota, M.; Grinshpun, B.; Sathaliyawala, T.; Kato, T.; Lerner, H.; Shen, Y.; Farber, D.L. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 2014, 159, 814–828. [Google Scholar] [CrossRef] [Green Version]
- Dock, J.; Ramirez, C.M.; Hultin, L.; Hausner, M.A.; Hultin, P.; Elliott, J.; Yang, O.O.; Anton, P.A.; Jamieson, B.D.; Effros, R.B. Distinct aging profiles of CD8+ T cells in blood versus gastrointestinal mucosal compartments. PLoS ONE 2017, 12, e0182498. [Google Scholar] [CrossRef] [PubMed]
- Martinet, K.Z.; Bloquet, S.; Bourgeois, C. Ageing combines CD4 T cell lymphopenia in secondary lymphoid organs and T cell accumulation in gut associated lymphoid tissue. Immun. Ageing 2014, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Booth, J.S.; Toapanta, F.R. B and T Cell Immunity in Tissues and Across the Ages. Vaccines 2021, 9, 24. https://doi.org/10.3390/vaccines9010024
Booth JS, Toapanta FR. B and T Cell Immunity in Tissues and Across the Ages. Vaccines. 2021; 9(1):24. https://doi.org/10.3390/vaccines9010024
Chicago/Turabian StyleBooth, Jayaum S., and Franklin R. Toapanta. 2021. "B and T Cell Immunity in Tissues and Across the Ages" Vaccines 9, no. 1: 24. https://doi.org/10.3390/vaccines9010024
APA StyleBooth, J. S., & Toapanta, F. R. (2021). B and T Cell Immunity in Tissues and Across the Ages. Vaccines, 9(1), 24. https://doi.org/10.3390/vaccines9010024