
A Bioinformatics Approach to Identifying Potential Biomarkers for Cryptosporidium parvum: A Coccidian Parasite Associated with Fetal Diarrhea

 , and
, and
Abstract
:1. Introduction
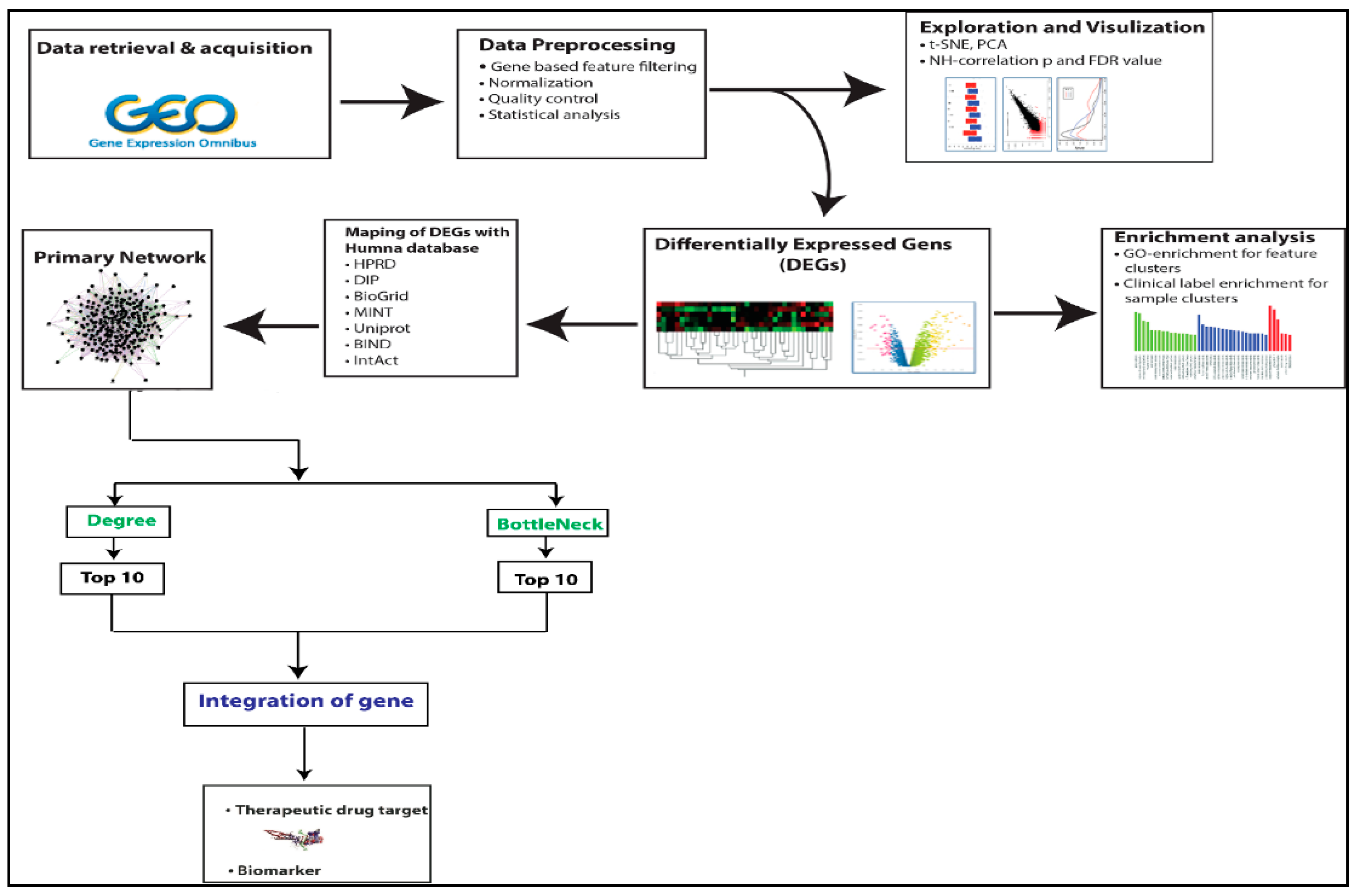
2. Method and Material
2.1. Microarray Datasets Collection and Pre-Processing
2.2. Identification of Differentially Expressed Genes (DEGs)
2.3. Construction and Analysis of PPI Networks
2.4. Topological Properties of the Network
2.5. Identification of Biomarker
2.6. Identification of miRNA Associated with Hub Genes
3. Results
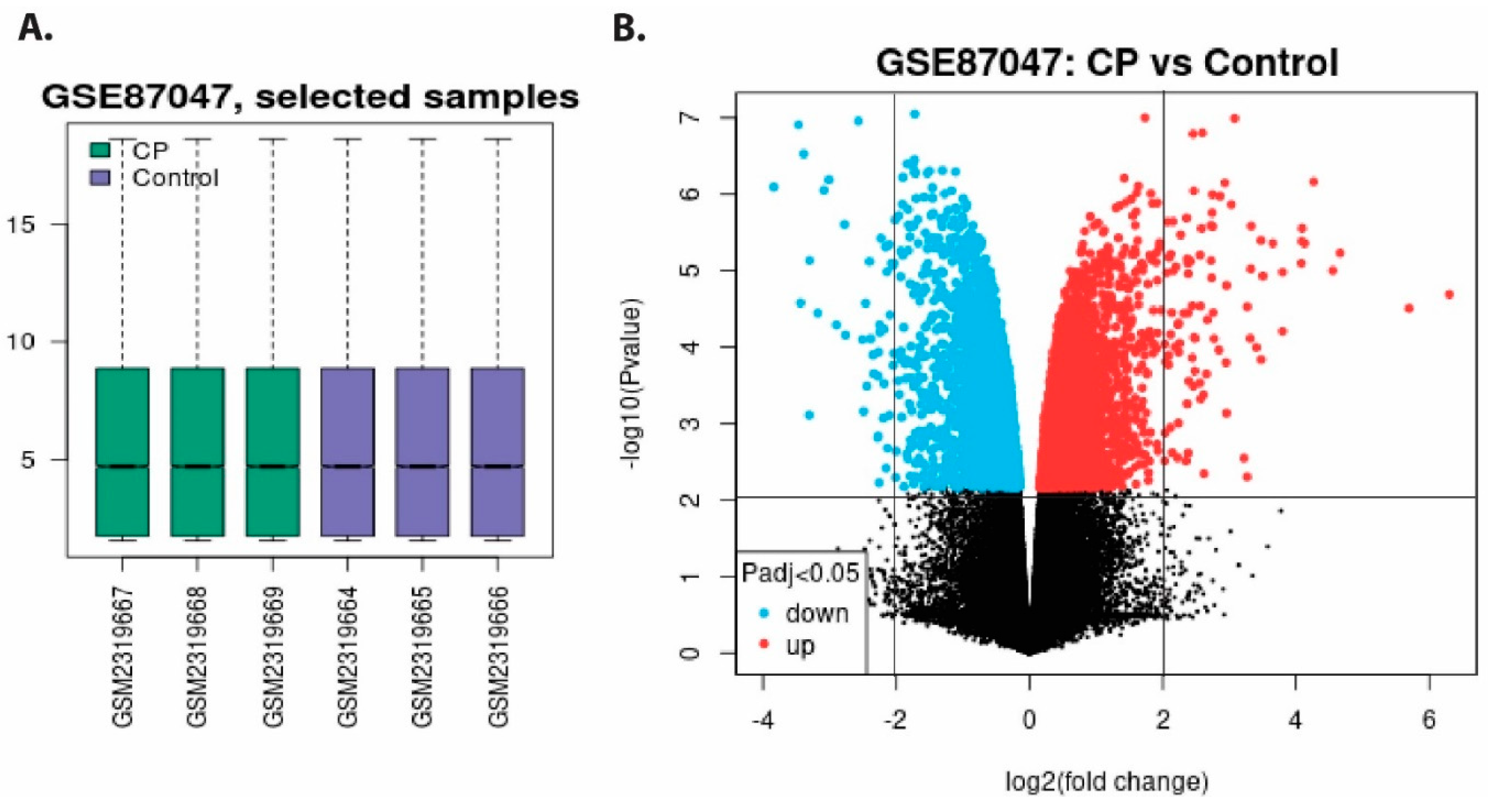
3.1. Extraction and Pre-Processing of Microarray Data
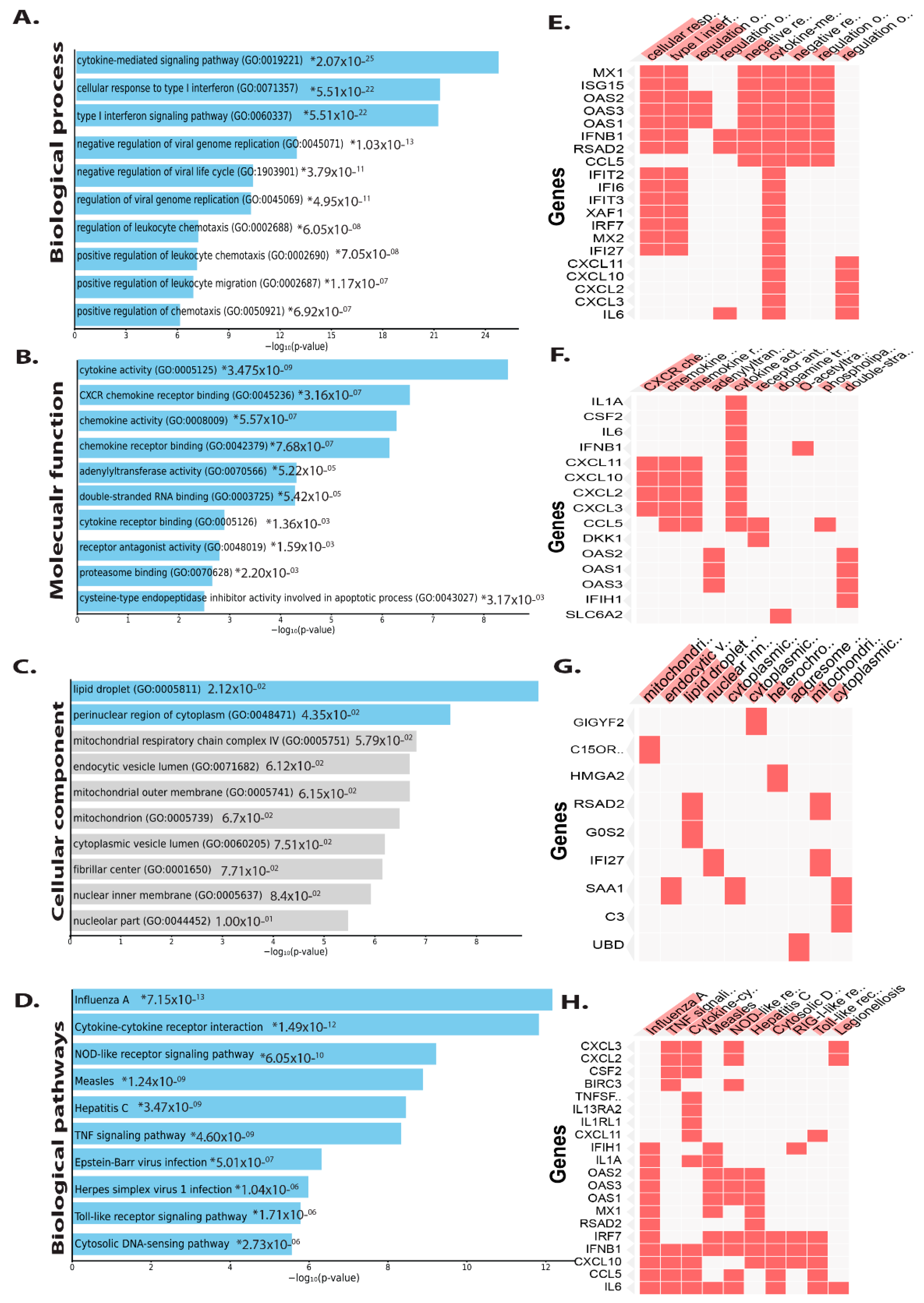
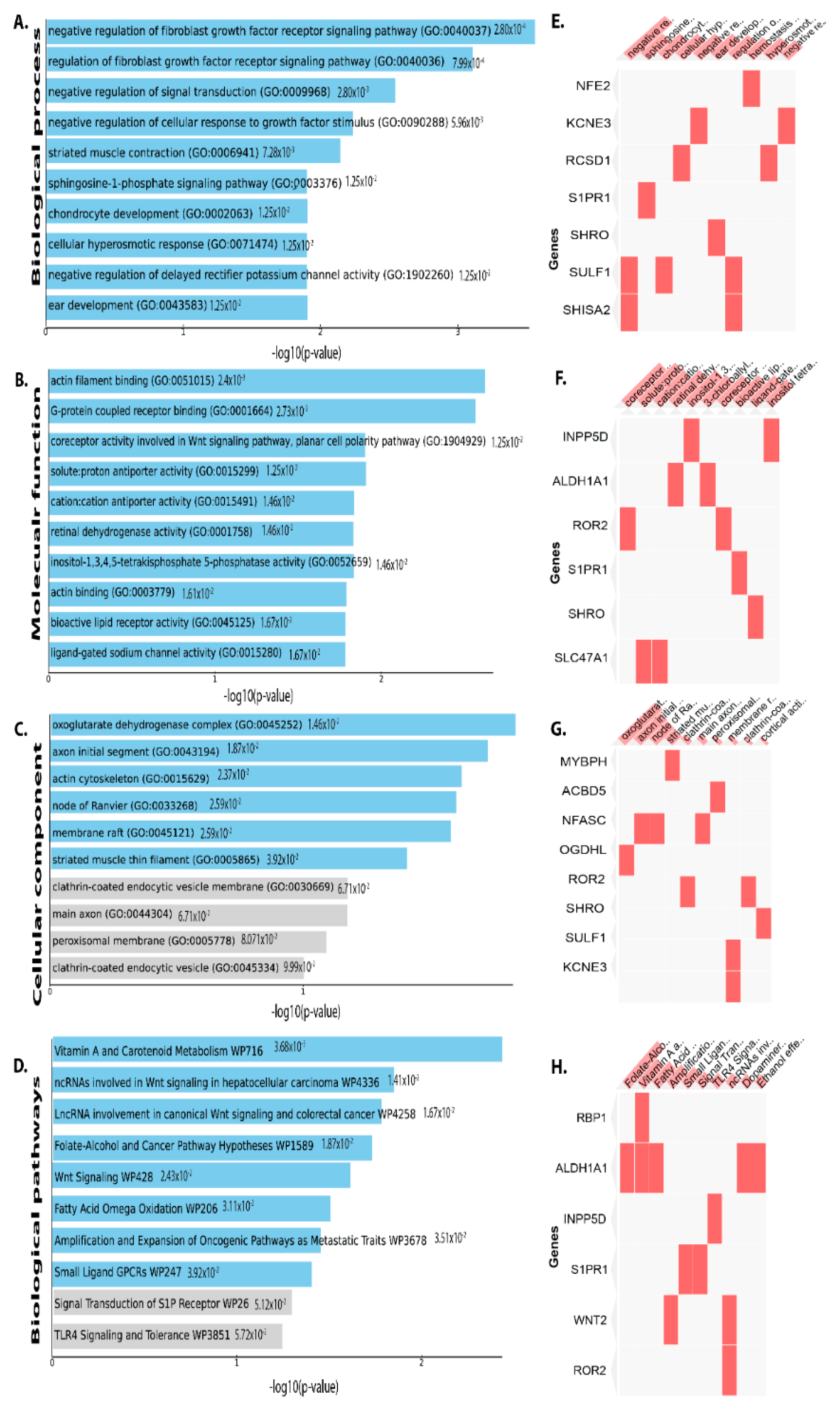
3.2. Functional and Pathway Enrichment Analysis
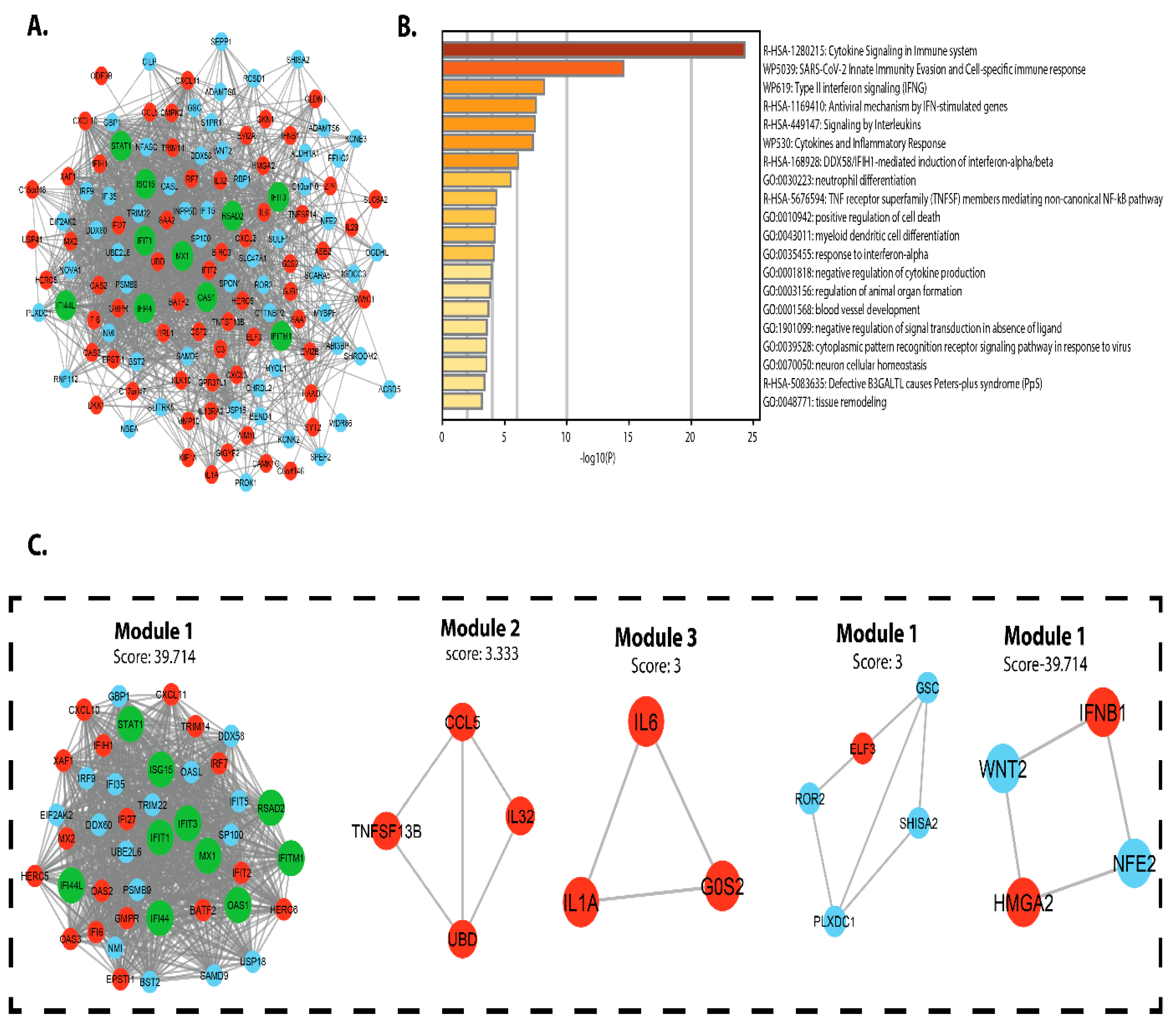
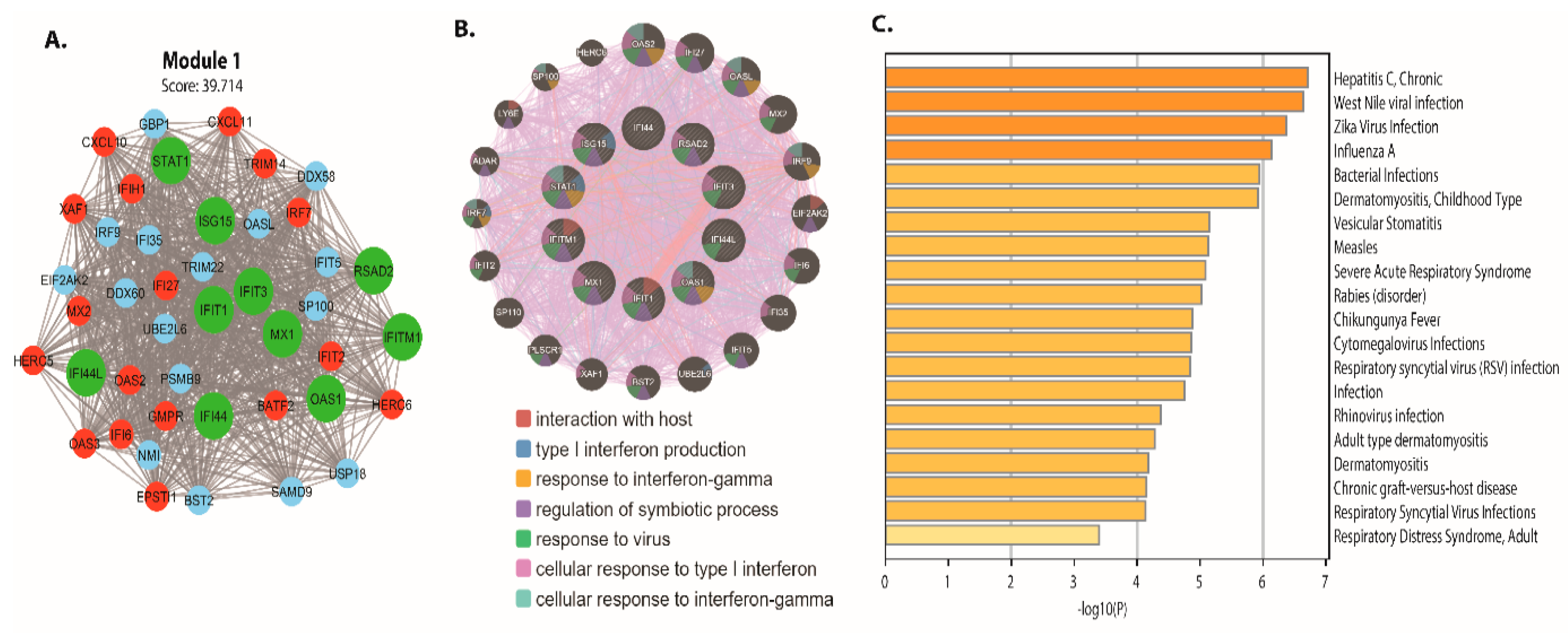
3.3. PPI Network and Module Analysis
3.4. Biomarker Identification
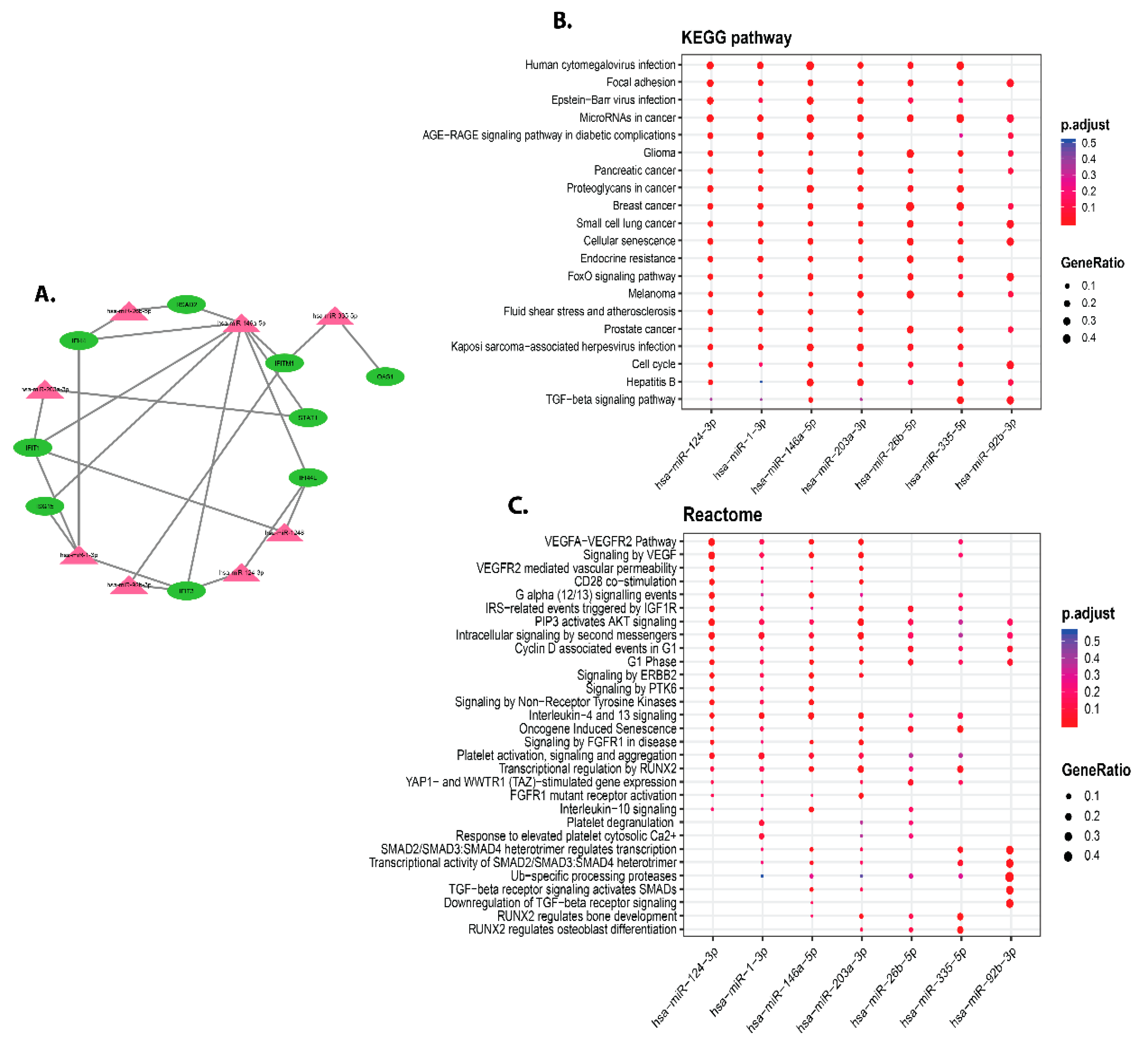
3.5. Identification of miRNAs Targeting Hub Genes of DEGs Infected by C. parvum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, A.; Shaik, J.S.; Grigg, M.E. Genomics and molecular epidemiology of Cryptosporidium species. Acta Trop. 2018, 184, 1–14. [Google Scholar] [CrossRef]
- Thomson, S.; Hamilton, C.A.; Hope, J.C.; Katzer, F.; Mabbott, N.A.; Morrison, L.J.; Innes, E.A. Bovine cryptosporidiosis: Impact, host-parasite interaction and control strategies. Vet. Res. 2017, 48, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomazic, M.L.; Maidana, J.; Dominguez, M.; Uriarte, E.L.; Galarza, R.; Garro, C.; Florin-Christensen, M.; Schnittger, L. Molecular characterization of Cryptosporidium isolates from calves in Argentina. Vet. Parasitol. 2013, 198, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.J.; Björkman, C.; Axén, C.; Fayer, R. Cryptosporidiosis in farmed animals. In Cryptosporidium: Parasite and Disease; Springer: Vienna, Austria, 2014; pp. 149–235. [Google Scholar]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Norman, F.F.; Monge-Maillo, B.; Martínez-Pérez, Á.; Perez-Molina, J.A.; López-Vélez, R. Parasitic infections in travelers and immigrants: Part I protozoa. Future Microbiol. 2015, 10, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Nime, F.A.; Burek, J.D.; Page, D.L.; Holscher, M.A.; Yardley, J.H. Acute enterocolitis in a human being infected with the protozoan Cryptosporidium. Gastroenterology 1976, 70, 592–598. [Google Scholar] [CrossRef]
- Bouzid, M.; Hunter, P.R.; Chalmers, R.M.; Tyler, K.M. Cryptosporidium pathogenicity and virulence. Clin. Microbiol. Rev. 2013, 26, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Feng, Y. Molecular epidemiologic tools for waterborne pathogens Cryptosporidium spp. and Giardia duodenalis. Food Waterborne Parasitol. 2017, 8, 14–32. [Google Scholar] [CrossRef]
- Yanta, C.A.; Bessonov, K.; Robinson, G.; Troell, K.; Guy, R.A. CryptoGenotyper: A new bioinformatics tool for rapid Cryptosporidium identification. Food Waterborne Parasitol. 2021, 23, e00115. [Google Scholar] [CrossRef]
- Janssen, B.; Snowden, J. Cryptosporidiosis; StatPearls Publishing: Treasure Island, FL, USA, 2017. [Google Scholar]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Burnet, J.-B.; Penny, C.; Ogorzaly, L.; Cauchie, H.-M. Spatial and temporal distribution of Cryptosporidium and Giardia in a drinking water resource: Implications for monitoring and risk assessment. Sci. Total Environ. 2014, 472, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Baldursson, S.; Karanis, P. Waterborne transmission of protozoan parasites: Review of worldwide outbreaks–an update 2004–2010. Water Res. 2011, 45, 6603–6614. [Google Scholar] [CrossRef]
- Chappell, C.L.; Okhuysen, P.C.; Langer-Curry, R.C.; Lupo, P.J.; Widmer, G.; Tzipori, S. Cryptosporidium muris: Infectivity and illness in healthy adult volunteers. Am. J. Trop. Med. Hyg. 2015, 92, 50. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Blom, U.M.; Wang, P.I.; Shim, J.E.; Marcotte, E.M. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011, 21, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.M.; Splinter, P.L.; O’Hara, S.P.; LaRusso, N.F. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J. Biol. Chem. 2007, 282, 28929–28938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A.J. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2010, 39, D1005–D1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Gong, A.Y.; Ma, S.; Chen, X.; Li, Y.; Su, C.J.; Norall, D.; Chen, J.; Strauss-Soukup, J.K.; Chen, X.M. Delivery of Parasite RNA Transcripts Into Infected Epithelial Cells During Cryptosporidium Infection and Its Potential Impact on Host Gene Transcription. J. Infect. Dis. 2017, 215, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Ming, Z.; Gong, A.Y.; Wang, Y.; Zhang, X.T.; Li, M.; Dolata, C.E.; Chen, X.M. Trans-suppression of defense DEFB1 gene in intestinal epithelial cells following Cryptosporidium parvum infection is associated with host delivery of parasite Cdg7_FLc_1000 RNA. Parasitol. Res. 2018, 117, 831–840. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, R.; Ahmed, M.M.; Tazyeen, S.; Alam, A.; Farooqui, A.; Ali, R.; Imam, N.; Tamkeen, N.; Ali, S.; Zubbair Malik, M. In Silico Integrative Approach Revealed Key MicroRNAs and Associated Target Genes in Cardiorenal Syndrome. Bioinform. Biol. Insights 2021, 15, 11779322211027396. [Google Scholar] [PubMed]
- Alam, A.; Imam, N.; Ahmed, M.M.; Tazyeen, S.; Tamkeen, N.; Farooqui, A.; Malik, M.; Ishrat, R. Identification and classification of differentially expressed genes and network meta-analysis reveals potential molecular signatures associated with tuberculosis. Front. Genet. 2019, 10, 932. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article3. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bandettini, W.P.; Kellman, P.; Mancini, C.; Booker, O.J.; Vasu, S.; Leung, S.W.; Wilson, J.R.; Shanbhag, S.M.; Chen, M.Y.; Arai, A.E. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: A clinical validation study. J. Cardiovasc. Magn. Reson. 2012, 14, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.-E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, M.Z.; Ali, S.; Singh, S.S.; Ishrat, R.; Singh, R.K.B. Dynamical states, possibilities and propagation of stress signal. Sci. Rep. 2017, 7, 40596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, M.Z.; Chirom, K.; Ali, S.; Ishrat, R.; Somvanshi, P.; Singh, R.K.B. Methodology of predicting novel key regulators in ovarian cancer network: A network theoretical approach. BMC Cancer 2019, 19, 1129. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Malik, M.Z.; Singh, S.S.; Chirom, K.; Ishrat, R.; Singh, R.K.B. Exploring novel key regulators in breast cancer network. PLoS ONE 2018, 13, e0198525. [Google Scholar] [CrossRef] [Green Version]
- Farooqui, A.; Tazyeen, S.; Ahmed, M.M.; Alam, A.; Ali, S.; Malik, M.Z.; Ali, S.; Ishrat, R. Assessment of the key regulatory genes and their Interologs for Turner Syndrome employing network approach. Sci. Rep. 2018, 8, 10091. [Google Scholar] [CrossRef] [PubMed]
- Mangangcha, I.R.; Malik, M.Z.; Kucuk, O.; Ali, S.; Singh, R.K.B. Kinless hubs are potential target genes in prostate cancer network. Genomics 2020, 112, 5227–5239. [Google Scholar] [CrossRef] [PubMed]
- Mangangcha, I.R.; Malik, M.Z.; Küçük, Ö.; Ali, S.; Singh, R.K.B. Identification of key regulators in prostate cancer from gene expression datasets of patients. Sci. Rep. 2019, 9, 16420. [Google Scholar] [CrossRef] [Green Version]
- Brandes, U. A faster algorithm for betweenness centrality. J. Math. Sociol. 2001, 25, 163–177. [Google Scholar] [CrossRef]
- Estrada, E.; Rodriguez-Velazquez, J.A. Subgraph centrality in complex networks. Phys. Rev. E 2005, 71, 056103. [Google Scholar] [CrossRef] [Green Version]
- Licursi, V.; Conte, F.; Fiscon, G.; Paci, P. MIENTURNET: An interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinform. 2019, 20, 545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Banaganapalli, B.; Al-Rayes, N.; Awan, Z.A.; Alsulaimany, F.A.; Alamri, A.S.; Elango, R.; Malik, M.Z.; Shaik, N.A. Multilevel Systems Biology Analysis of Lung Transcriptomics Data Identifies key miRNAs and Potential miRNA Target Genes for SARS-CoV-2 Infection. Comput. Biol. Med. 2021, 135, 104570. [Google Scholar]
- Gupta, V.; Haider, S.; Verma, M.; Singhvi, N.; Ponnusamy, K.; Malik, M.Z.; Verma, H.; Kumar, R.; Sood, U.; Hira, P.; et al. Comparative Genomics and Integrated Network Approach Unveiled Undirected Phylogeny Patterns, Co-mutational Hotspots, Functional Crosstalk and Regulatory Interactions in SARS-CoV-2. MSystems 2021, 6, e00030-21. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; AlOmar, S.Y.; Alqahtani, S.A.M.; Malik, Z.; Kumar, V. Brain Disease Network analysis to elucidate the neurological manifestations of COVID-19. Mol. Neurobiol. 2021, 58, 1875–1893. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.C.; Mac-Kenzie, W.R.; Addiss, D.G.; Gradus, M.S.; Linke, G.; Zembrowski, E. Cryptosporidium parvum-specific antibody responses among children residing in Milwaukee during the 1993 waterborne outbreak. J. Infect. Dis. 2001, 183, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Harwood, M.; Girouard, D.; Meyers, M.J.; Campbell, M.A.; Beamer, G. The therapeutic efficacy of azithromycin and nitazoxanide in the acute pig model of Cryptosporidium hominis. PLoS ONE 2017, 12, e0185906. [Google Scholar] [CrossRef] [Green Version]
- Atianand, M.K.; Fitzgerald, K.A. Long non-coding RNAs and control of gene expression in the immune system. Trends Mol. Med. 2014, 20, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-M.; Gores, G.J.; Paya, C.V.; LaRusso, N.F. Cryptosporidium parvum induces apoptosis in biliary epithelia by a Fas/Fas ligand-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 1999, 277, G599–G608. [Google Scholar] [CrossRef]
- Ojcius, D.M.; Perfettini, J.L.; Bonnin, A.; Laurent, F. Caspase-dependent apoptosis during infection with Cryptosporidium parvum. Microbes Infect. 1999, 1, 1163–1168. [Google Scholar] [CrossRef]
- McCole, D.F.; Eckmann, L.; Laurent, F.; Kagnoff, M.F. Intestinal epithelial cell apoptosis following Cryptosporidium parvum infection. Infect. Immun. 2000, 68, 1710–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-L.; Buck, G.A.; Widmer, G. Cell sorting-assisted microarray profiling of host cell response to Cryptosporidium parvum infection. Infect. Immun. 2010, 78, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Prasanth, K.V.; Spector, D.L. Eukaryotic regulatory RNAs: An answer to the ‘genome complexity’conundrum. Genes Dev. 2007, 21, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Puiu, D.; Enomoto, S.; Buck, G.A.; Abrahamsen, M.S.; Kissinger, J.C. CryptoDB: The Cryptosporidium genome resource. Nucleic Acids Res. 2004, 32, D329–D331. [Google Scholar] [CrossRef]
- Vembar, S.S.; Scherf, A.; Siegel, T.N. Noncoding RNAs as emerging regulators of Plasmodium falciparum virulence gene expression. Curr. Opin. Microbiol. 2014, 20, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamsen, M.S.; Templeton, T.J.; Enomoto, S.; Abrahante, J.E.; Zhu, G.; Lancto, C.A.; Deng, M.; Liu, C.; Widmer, G.; Tzipori, S. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 2004, 304, 441–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Q.; Shen, J.; Liu, J.; Sun, X.; Zhao, G.; Chang, Y.; Xu, L.; Li, X.; Zhao, Y.; Zheng, H. Genome-wide identification and functional annotation of Plasmodium falciparum long noncoding RNAs from RNA-seq data. Parasitol. Res. 2014, 113, 1269–1281. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, J.; Wakaguri, H.; Sugano, S.; Kawano, S.; Fujisaki, K.; Sugimoto, C.; Watanabe, J.; Suzuki, Y.; Kimata, I.; Xuan, X. Construction and analysis of full-length cDNA library of Cryptosporidium parvum. Parasitol. Int. 2011, 60, 199–202. [Google Scholar] [CrossRef]
- Liu, X.; Lu, R.; Xia, Y.; Sun, J. Global analysis of the eukaryotic pathways and networks regulated by Salmonella typhimurium in mouse intestinal infection in vivo. BMC Genom. 2010, 11, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shtrichman, R.; Heithoff, D.M.; Mahan, M.J.; Samuel, C.E. Tissue selectivity of interferon-stimulated gene expression in mice infected with Dam+ versus Dam− Salmonella enterica serovar Typhimurium strains. Infect. Immun. 2002, 70, 5579–5588. [Google Scholar] [CrossRef] [Green Version]
- West, K.O.; Wagner, A.R.; Scott, H.M.; Vail, K.J.; Carter, K.E.; Watson, R.O.; Patrick, K.L. Global transcriptomics reveals specialized roles for splicing regulatory proteins in the macrophage innate immune response. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sibley, L.D. Intracellular parasite invasion strategies. Science 2004, 304, 248–253. [Google Scholar] [CrossRef]
- O’Connor, R.M.; Wanyiri, J.W.; Wojczyk, B.S.; Kim, K.; Ward, H. Stable expression of Cryptosporidium parvum glycoprotein gp40/15 in Toxoplasma gondii. Mol. Biochem. Parasitol. 2007, 152, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gong, A.Y.; Ma, S.; Chen, X.; Strauss-Soukup, J.K.; Chen, X.M. Delivery of parasite Cdg7_Flc_0990 RNA transcript into intestinal epithelial cells during Cryptosporidium parvum infection suppresses host cell gene transcription through epigenetic mechanisms. Cell. Microbiol. 2017, 19, e12760. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Hu, G.; Liu, J.; Gong, A.-Y.; Drescher, K.M.; Chen, X.-M. NF-kappaB p65-dependent transactivation of miRNA genes following Cryptosporidium parvum infection stimulates epithelial cell immune responses. PLoS Pathog. 2009, 5, e1000681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Gong, A.-Y.; Eischeid, A.N.; Chen, X.-M. miR-27b targets KSRP to coordinate TLR4-mediated epithelial defense against Cryptosporidium parvum infection. PLoS Pathog. 2012, 8, e1002702. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Koshizuka, T.; Ishibashi, K.; Hashimoto, K.; Ishioka, K.; Ikuta, K.; Yokota, S.i.; Fujii, N.; Suzutani, T. Involvement of herpes simplex virus type 1 UL13 protein kinase in induction of SOCS genes, the negative regulators of cytokine signaling. Microbiol. Immunol. 2017, 61, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tan, S.; Kooger, R.; Zhang, C.; Zhang, Y.J.C.S.R. MicroRNAs as novel biological targets for detection and regulation. Chem. Soc. Rev. 2014, 43, 506–517. [Google Scholar] [CrossRef]
- Curtale, G.; Mirolo, M.; Renzi, T.A.; Rossato, M.; Bazzoni, F.; Locati, M. Negative regulation of Toll-like receptor 4 signaling by IL-10–dependent microRNA-146b. Proc. Natl. Acad. Sci. USA 2013, 110, 11499–11504. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Genes | Degree | Gene | BottleNeck |
|---|---|---|---|---|
| 1. | ISG15 | 355 | IFIT3 | 62 |
| 2. | MX1 | 349 | IFITM1 | 58 |
| 3. | IFI44L | 291 | GBP1 | 58 |
| 4. | STAT1 | 286 | IFI44 | 56 |
| 5. | IFIT1 | 283 | IFIT2 | 56 |
| 6. | OAS1 | 280 | MX1 | 54 |
| 7. | IFIT3 | 280 | TRIM22 | 54 |
| 8. | RSAD2 | 263 | NMI | 54 |
| 9. | IFITM1 | 258 | IFI27 | 54 |
| 10. | IFI44 | 251 | IFIT5 | 53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabir, M.J.; Low, R.; Hall, N.; Kamli, M.R.; Malik, M.Z. A Bioinformatics Approach to Identifying Potential Biomarkers for Cryptosporidium parvum: A Coccidian Parasite Associated with Fetal Diarrhea. Vaccines 2021, 9, 1427. https://doi.org/10.3390/vaccines9121427
Sabir MJ, Low R, Hall N, Kamli MR, Malik MZ. A Bioinformatics Approach to Identifying Potential Biomarkers for Cryptosporidium parvum: A Coccidian Parasite Associated with Fetal Diarrhea. Vaccines. 2021; 9(12):1427. https://doi.org/10.3390/vaccines9121427
Chicago/Turabian StyleSabir, Mumdooh J., Ross Low, Neil Hall, Majid Rasool Kamli, and Md. Zubbair Malik. 2021. "A Bioinformatics Approach to Identifying Potential Biomarkers for Cryptosporidium parvum: A Coccidian Parasite Associated with Fetal Diarrhea" Vaccines 9, no. 12: 1427. https://doi.org/10.3390/vaccines9121427
APA StyleSabir, M. J., Low, R., Hall, N., Kamli, M. R., & Malik, M. Z. (2021). A Bioinformatics Approach to Identifying Potential Biomarkers for Cryptosporidium parvum: A Coccidian Parasite Associated with Fetal Diarrhea. Vaccines, 9(12), 1427. https://doi.org/10.3390/vaccines9121427