
Immunoinformatics Analysis of SARS-CoV-2 ORF1ab Polyproteins to Identify Promiscuous and Highly Conserved T-Cell Epitopes to Formulate Vaccine for Indonesia and the World Population



Abstract
:1. Introduction
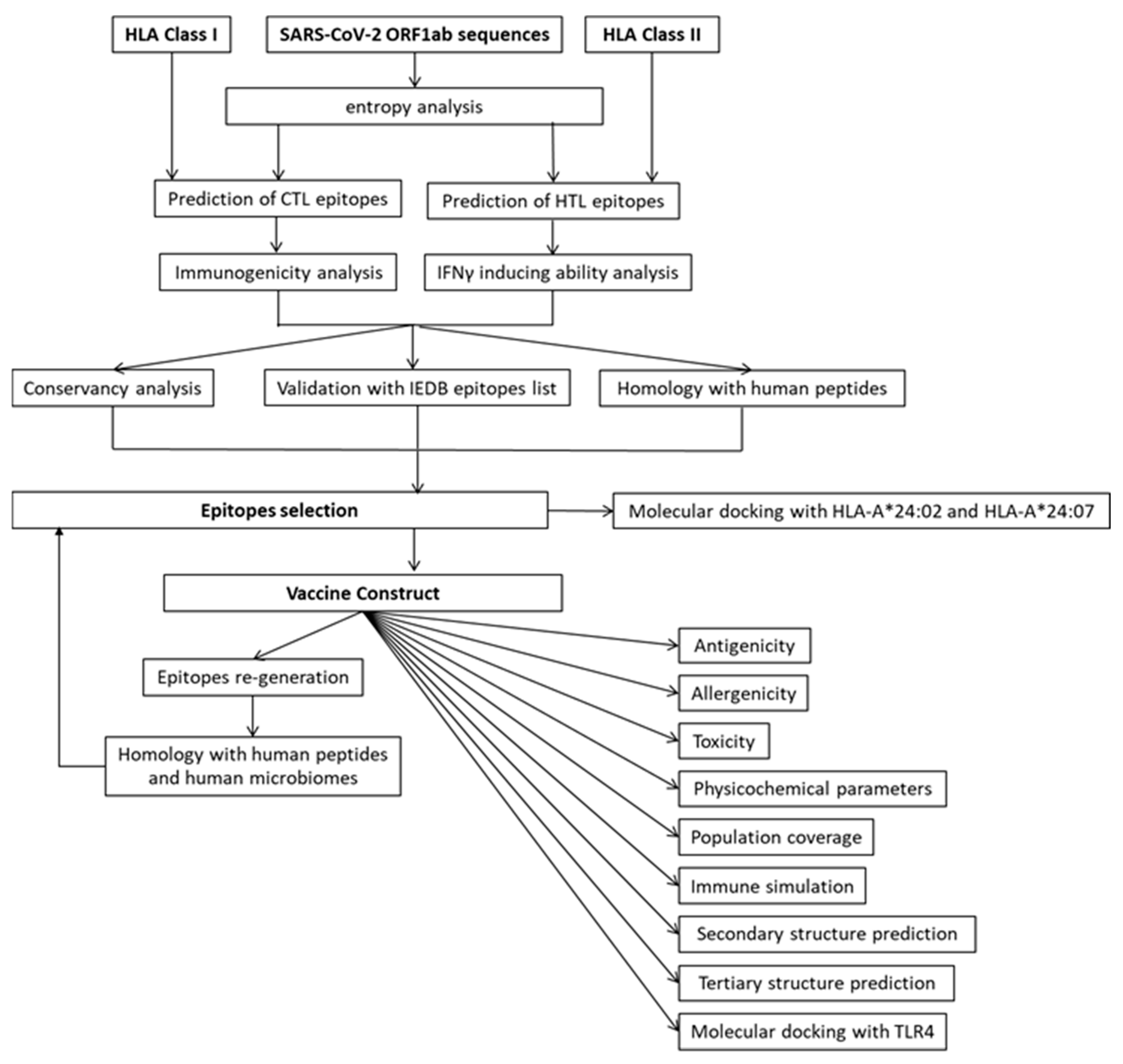
2. Materials and Methods
2.1. SARS-CoV-2 ORF1ab Sequence Retrieval
2.2. Entropy Analysis of 9-Mer Peptide Sequences
2.3. Retrieval of HLA Alleles Type in INDONESIAN Population as the Bases for Prediction
2.4. Retrieval of the Number of Experimentally Validated ORF1ab Epitopes Associated with Predominant Indonesian HLA Alleles
2.5. Prediction of CTL Epitopes from ORF1ab
2.6. Prediction of HTL Epitopes from ORF1ab
2.7. Immunogenicity Analysis of Predicted CTL Epitopes
2.8. Interferon-Gamma (IFNγ)-Inducing Ability of Predicted HTL Epitopes
2.9. Conservancy Analysis of the Predicted Epitopes against SARS-CoV-2 Variants
2.10. Validation of Predicted Epitopes in IEDB Epitopes List
2.11. Cross-Reactivity of Predicted Epitopes with Human Peptides
2.12. Epitope Selection and Vaccine Construction
2.13. Evaluation of VC Properties: Antigenicity, Allergenicity, Toxicity, and Physicochemical Characteristics
2.14. Re-Analyze the VC for Epitopes Generation and Homology with Human Proteins and Human Microbiome
2.15. Immune Simulation of the VC
2.16. Population Coverage of the VC
2.17. Secondary Structure and Tertiary Structure Prediction of the VC
2.18. Molecular Docking of the VC with TLR4
2.19. Molecular Docking of Peptide WSMATYYLF with HLA-A*24:02 and HLA-A*24:07
3. Results
3.1. SARS-CoV-2 ORF1ab Polyprotein Contains Evolutionary Stable Regions with Low Entropy
3.2. SARS-CoV-2 ORF1ab Contributes a Large Number of Experimentally Known Immunogenic Epitopes in IEDB
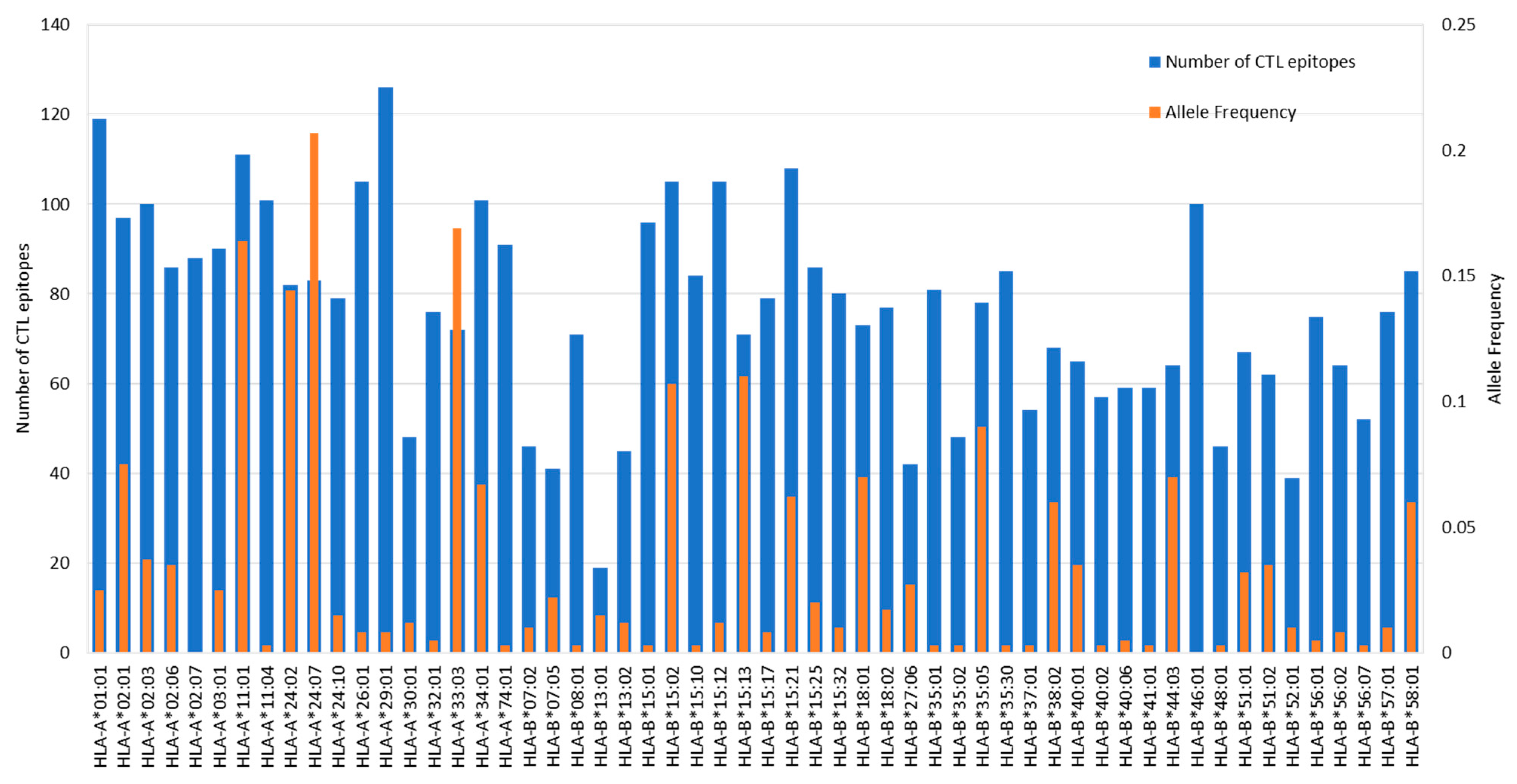
3.3. HLA Allele Frequencies of the Indonesian, Thai, and German Population
3.4. Asian HLA Alleles Are Less Studied as Compared to the HLA Alleles Predominant in the European Population
3.5. Prediction of CTL Epitopes and Evaluation of Immunogenicity
3.6. Prediction of HTL Epitopes and Evaluation of IFNγ Induction Capability
3.7. Conservancy Analysis
3.8. Comparison of Predicted Epitopes and Experimentally Proven Epitopes from IEDB
3.9. Homology with Human Peptides
3.10. Epitope Cross-Reactivity with Human Peptides, Human Common Cold Coronaviruses (HCCs), or Other Ubiquitous Antigens
3.11. Epitope Selection
3.12. Population Coverage
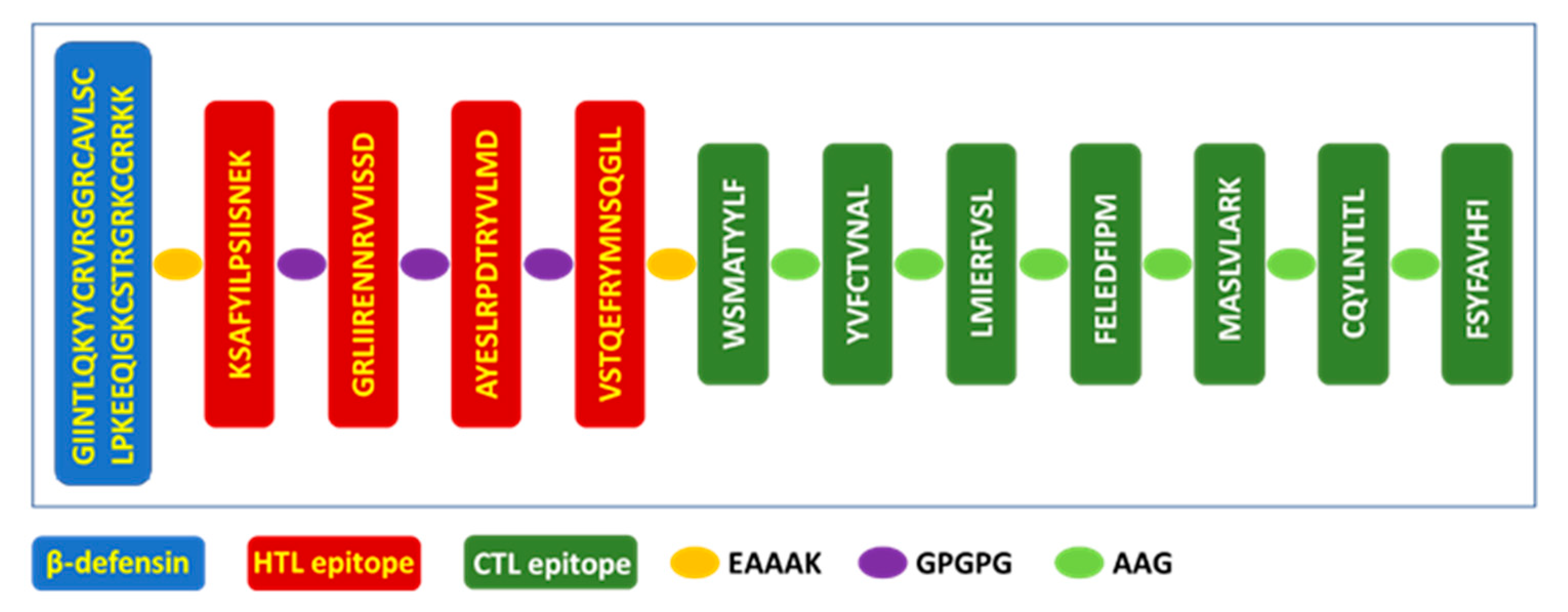
3.13. Vaccine Design
3.14. Vaccine Antigenicity, Allergenicity, Toxicity, and Physicochemical Characteristics
3.15. Re-Analyze the VC for Epitopes Generation and Homology with Human Proteins and Microbiomes
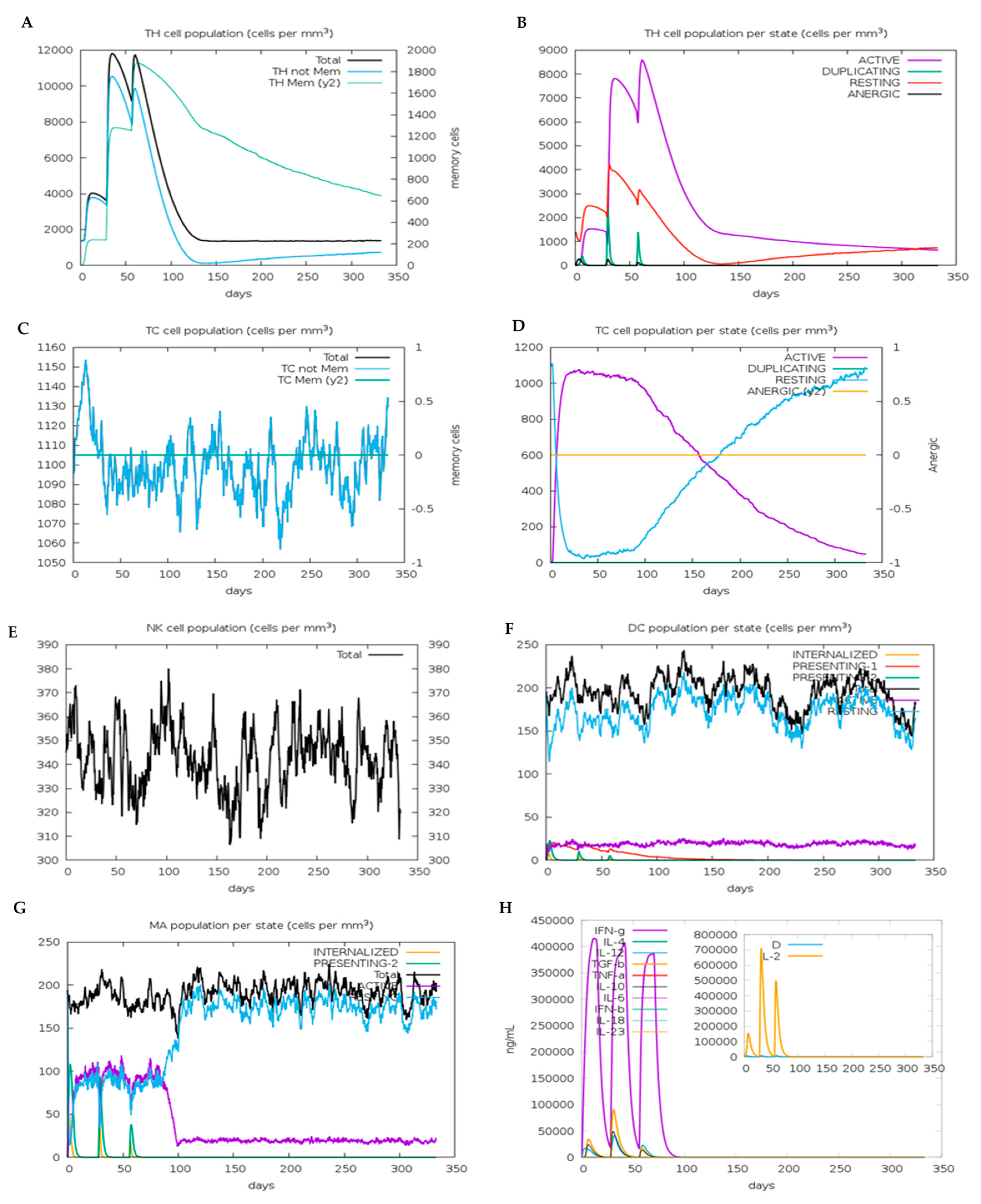
3.16. In Silico Immune Simulation of the VC
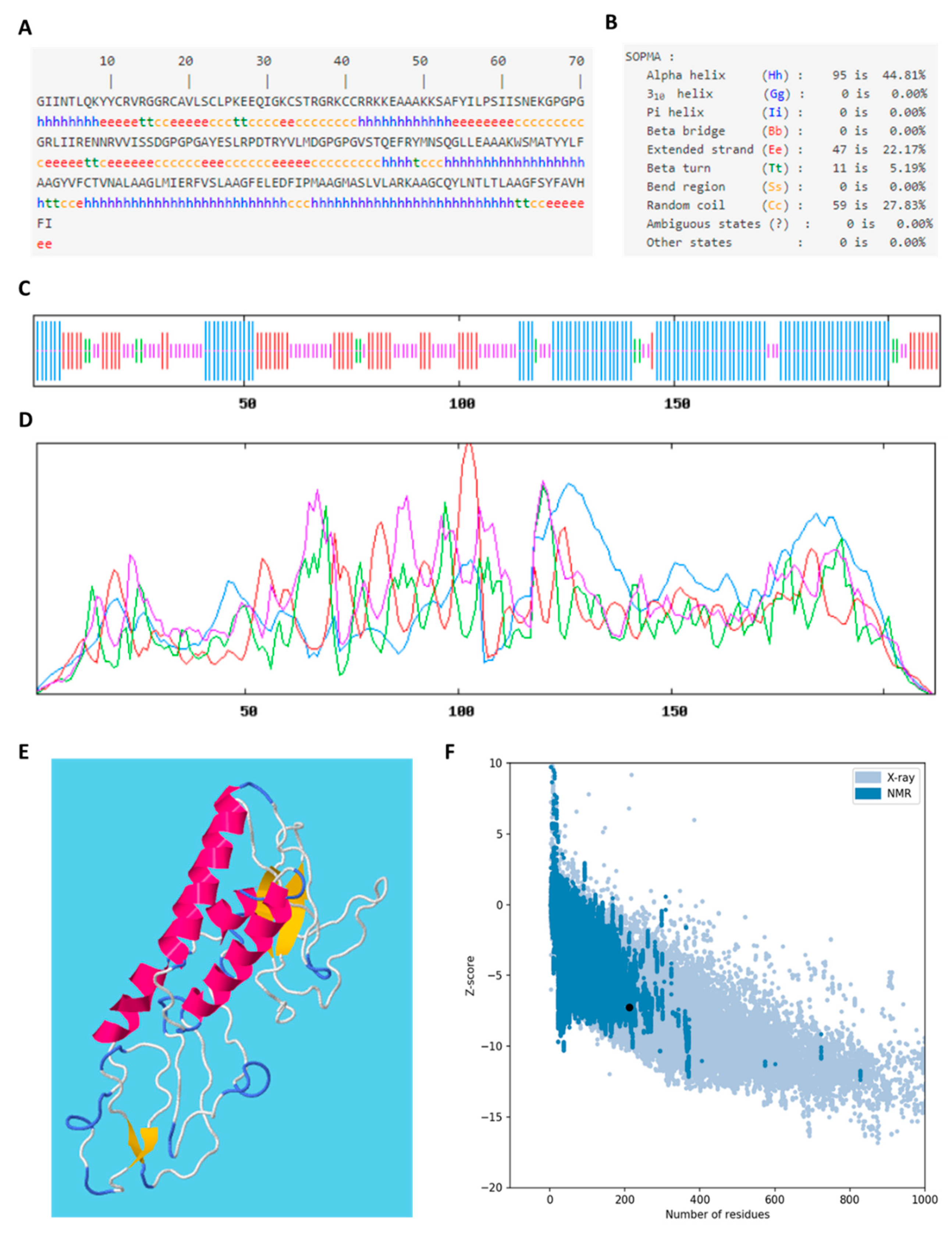
3.17. Secondary Structure and Tertiary Structure of Vaccine Construct
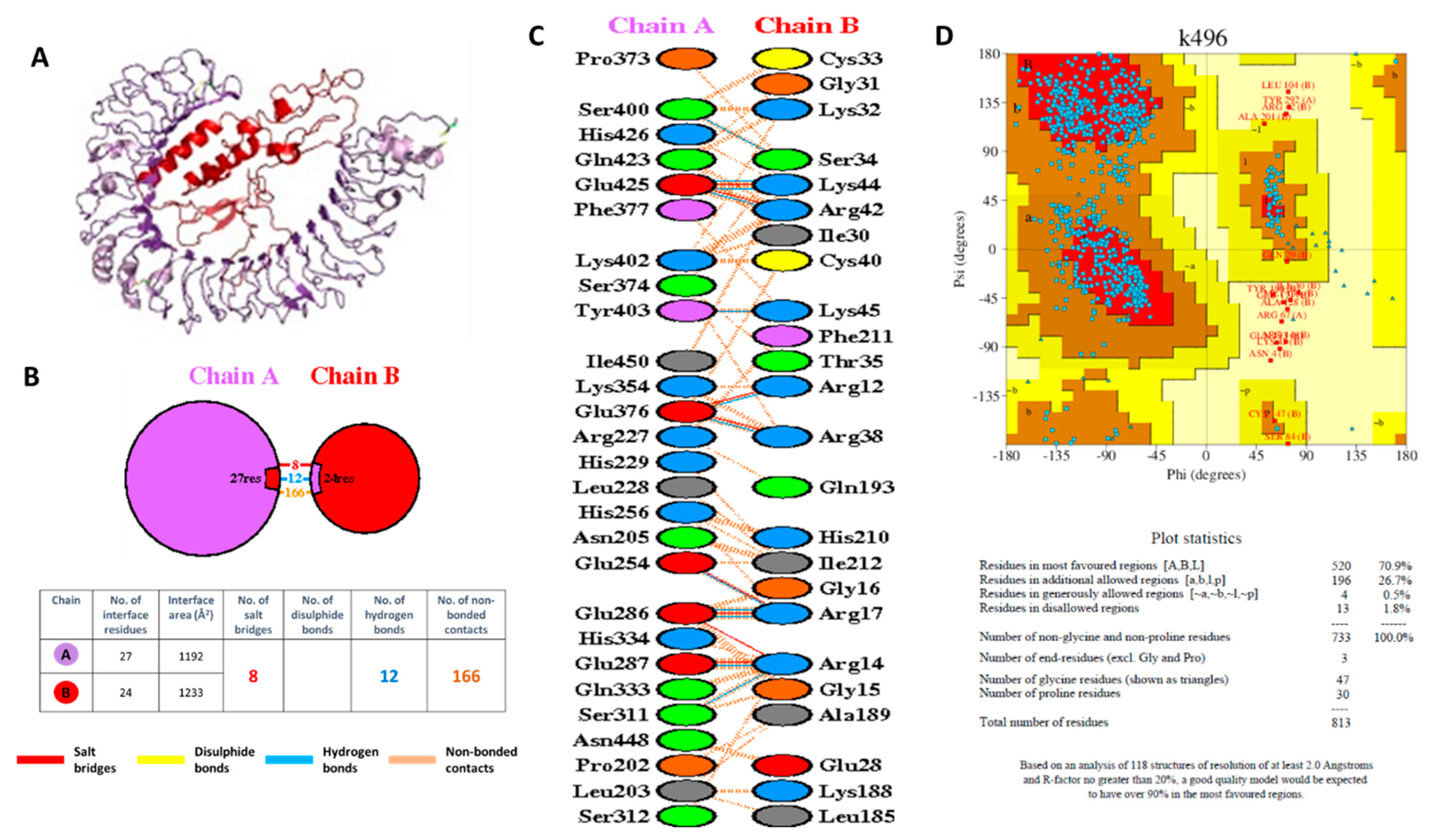
3.18. Molecular Docking of the VC with TLR4
3.19. Molecular Docking Simulation of Peptide Binding to HLA-A*24:02 and HLA-A*24:07
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, S.; Siddique, R.; Shereen, M.A.; Ali, A.; Liu, J.; Bai, Q.; Bashir, N.; Xue, M. Emergence of a Novel Coronavirus, Severe Acute Respiratory Syndrome Coronavirus 2: Biology and Therapeutic Options. J. Clin. Microbiol. 2020, 58, e00187-20. [Google Scholar] [CrossRef] [Green Version]
- Mo, P.; Xing, Y.; Xiao, Y.; Deng, L.; Zhao, Q.; Wang, H.; Xiong, Y.; Cheng, Z.; Gao, S.; Liang, K.; et al. Clinical Characteristics of Refractory COVID-19 Pneumonia in Wuhan, China. Clin. Infect. Dis. 2020, ciaa270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 21 September 2021).
- Satuan Tugas Penanganan COVID-19 Peta Sebaran COVID-19. Available online: https://covid19.go.id/peta-sebaran (accessed on 26 September 2021).
- Zhou, P.; Yang, L.-X.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19) (Updated 2021 Sep 2). In StatPearls. Treasure Island (FL): StatPearls Publishing; 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554776/ (accessed on 2 September 2021).
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging Coronaviruses: Genome Structure, Replication, and Pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Mlcochova, P.; Kemp, S.A.; Dhar, M.S.; Papa, G.; Meng, B.; Ferreira, I.A.T.M.; Datir, R.; Collier, D.A.; Albecka, A.; Singh, S.; et al. SARS-CoV-2 B.1.617.2 Delta Variant Replication and Immune Evasion. Nature 2021, 599, 114–119. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 Genomic Variations Associated with Mortality Rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 23 September 2021).
- World Health Organization. COVID-19 Vaccine Tracker and Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-COVID-19-candidate-vaccines (accessed on 24 September 2021).
- Vilar, S.; Isom, D.G. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef]
- Khan, A.M.; Hu, Y.; Miotto, O.; Thevasagayam, N.M.; Sukumaran, R.; Abd Raman, H.S.; Brusic, V.; Tan, T.W.; Thomas August, J. Analysis of Viral Diversity for Vaccine Target Discovery. BMC Med. Genom. 2017, 10, 78–92. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Miotto, O.; Heiny, A.; Tan, T.W.; August, J.T.; Brusic, V. Identification of Human-to-Human Transmissibility Factors in PB2 Proteins of Influenza A by Large-Scale Mutual Information Analysis. BMC Bioinform. 2008, 9, S18. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Galarza, F.F.; McCabe, A.; Santos, E.J.; Jones, J.; Takeshita, L.Y.; Ortega-Rivera, N.D.; del Cid-Pavon, G.M.; Ramsbottom, K.; Ghattaoraya, G.S.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acid Res. 2020, 48, D783–D788. [Google Scholar] [CrossRef]
- Yuliwulandari, R.; Kashiwase, K.; Nakajima, H.; Uddin, J.; Susmiarsih, T.P.; Sofro, A.S.M.; Tokunaga, K. Polymorphisms of HLA Genes in Western Javanese (Indonesia): Close Affinities to Southeast Asian Populations. Tissue Antigens 2009, 73, 46–53. [Google Scholar] [CrossRef] [PubMed]
- World Population Review. Indonesia Population. Available online: https://worldpopulationreview.com/countries/indonesia-population (accessed on 1 October 2021).
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelde, A.; Bilich, T.; Heitmann, J.; Maringer, Y.; Salih, H.; Roerden, M.; Lübke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition. Nat. Immunol. 2021, 22, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Geretz, A.; Ehrenberg, P.K.; Bouckenooghe, A.; Fernández Viña, M.A.; Michael, N.L.; Chansinghakule, D.; Limkittikul, K.; Thomas, R. Full-Length next-Generation Sequencing of HLA Class I and II Genes in a Cohort from Thailand. Hum. Immunol. 2018, 79, 773–780. [Google Scholar] [CrossRef]
- Prentice, H.A.; Ehrenberg, P.K.; Baldwin, K.M.; Geretz, A.; Andrews, C.; Nitayaphan, S.; Rerks-Ngarm, S.; Kaewkungwal, J.; Pitisuttithum, P.; O’Connell, R.J.; et al. HLA Class I, KIR, and Genome-Wide SNP Diversity in the RV144 Thai Phase 3 HIV Vaccine Clinical Trial. Immunogenetics 2014, 66, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.M.; Ehrenberg, P.K.; Geretz, A.; Prentice, H.A.; Nitayaphan, S.; Rerks-Ngarm, S.; Kaewkungwal, J.; Pitisuttithum, P.; O’Connell, R.J.; Kim, J.H.; et al. HLA Class II Diversity in HIV-1 Uninfected Individuals from the Placebo Arm of the RV144 Thai Vaccine Efficacy Trial. Tissue Antigens 2015, 85, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stranzl, T.; Larsen, M.V.; Lundegaard, C.; Nielsen, M. NetCTLpan: Pan-Specific MHC Class I Pathway Epitope Predictions. Immunogenetics 2010, 62, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backert, L.; Kohlbacher, O. Immunoinformatics and Epitope Prediction in the Age of Genomic Medicine. Genome Med. 2015, 7, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Reynisson, B.; Barra, C.; Kaabinejadian, S.; Hildebrand, W.H.; Peters, B.; Nielsen, M. Improved Prediction of MHC II Antigen Presentation through Integration and Motif Deconvolution of Mass Spectrometry MHC Eluted Ligand Data. J. Proteome Res. 2020, 19, 2304–2315. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.A.; de Boer, R.J.; Keşmir, C. Degenerate T-Cell Recognition of Peptides on MHC Molecules Creates Large Holes in the T-Cell Repertoire. PLoS Comput. Biol. 2012, 8, e1002412. [Google Scholar] [CrossRef] [Green Version]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; de Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLoS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of Interferon-Gamma Inducing MHC Class-II Binders. Biol. Direct 2013, 8, 30–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.H.; Sidney, J.; Li, W.; Fusseder, N.; Sette, A. Development of an Epitope Conservancy Analysis Tool to Facilitate the Design of Epitope-Based Diagnostics and Vaccines. BMC Bioinform. 2007, 8, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Cuspoca, A.F.; Díaz, L.L.; Acosta, A.F.; Peñaloza, M.K.; Méndez, Y.R.; Clavijo, D.C.; Reyes, J.Y. An Immunoinformatics Approach for SARS-CoV-2 in Latam Populations and Multi-Epitope Vaccine Candidate Directed towards the World’s Population. Vaccines 2021, 9, 581. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A Server for in Silico Prediction of Allergens. BMC Bioinform. 2013, 14, S4–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity Prediction by Descriptor Fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Analysis Tools on the ExPASy Server 571 571 From: The Proteomics Protocols Handbook Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco Pro, S.; Lindestam Arlehamn, C.S.; Dhanda, S.K.; Carpenter, C.; Lindvall, M.; Faruqi, A.A.; Santee, C.A.; Renz, H.; Sidney, J.; Peters, B.; et al. Microbiota Epitope Similarity Either Dampens or Enhances the Immunogenicity of Disease-Associated Antigenic Epitopes. PLoS ONE 2018, 13, e0196551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shende, G.; Haldankar, H.; Barai, R.S.; Bharmal, M.H.; Shetty, V.; Idicula-Thomas, S. PBIT: Pipeline Builder for Identification of Drug Targets for Infectious Diseases. Bioinformatics 2016, 33, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational Immunology Meets Bioinformatics: The Use of Prediction Tools for Molecular Binding in the Simulation of the Immune System. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.S.; Hossan, M.I.; Mizan, S.; Moin, A.T.; Yasmin, F.; Akash, A.-S.; Powshi, S.N.; Hasan, A.K.R.; Chowdhury, A.S. Immunoinformatics Approach to Designing a Multi-Epitope Vaccine against Saint Louis Encephalitis Virus. Inform. Med. Unlocked 2021, 22, 100500. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate De Novo Prediction of Protein Contact Map by Ultra-Deep Learning Model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef] [Green Version]
- Xu, J. Distance-Based Protein Folding Powered by Deep Learning. Proc. Natl. Acad. Sci. USA 2019, 116, 16856–16865. [Google Scholar] [CrossRef] [Green Version]
- Sippl, M.J. Recognition of Errors in Three-Dimensional Structures of Proteins. Proteins Struct. Funct. Genet. 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK Server for Integrated Protein–Protein Docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A Web Server for Protein–Protein and Protein–DNA/RNA Docking Based on a Hybrid Strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro Web Server for Protein–Protein Docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum New Things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef]
- Blaszczyk, M.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. Protein–Peptide Docking Using CABS-Dock and Contact Information. Brief. Bioinform. 2019, 20, 2299–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurcinski, M.; Blaszczyk, M.; Ciemny, M.P.; Kolinski, A.; Kmiecik, S. A Protocol for CABS-Dock Protein–Peptide Docking Driven by Side-Chain Contact Information. BioMedical Eng. Online 2017, 16, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaszczyk, M.; Kurcinski, M.; Kouza, M.; Wieteska, L.; Debinski, A.; Kolinski, A.; Kmiecik, S. Modeling of Protein–Peptide Interactions Using the CABS-Dock Web Server for Binding Site Search and Flexible Docking. Methods 2016, 93, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Mullick, B.; Magar, R.; Jhunjhunwala, A.; Barati Farimani, A. Understanding Mutation Hotspots for the SARS-CoV-2 Spike Protein Using Shannon Entropy and K-Means Clustering. Comput. Biol. Med. 2021, 138, 104915. [Google Scholar] [CrossRef]
- Thomas, S. Mapping the Nonstructural Transmembrane Proteins of Severe Acute Respiratory Syndrome Coronavirus 2. J. Comput. Biol. 2021, 28, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Shcherbik, N.; Sawaya, B.E. Why Do SARS-CoV-2 NSPs Rush to the ER? J. Neurol. 2021, 268, 2013–2022. [Google Scholar] [CrossRef] [PubMed]
- Gorkhali, R.; Koirala, P.; Rijal, S.; Mainali, A.; Baral, A.; Bhattarai, H.K. Structure and Function of Major SARS-CoV-2 and SARS-CoV Proteins. Bioinform. Biol. Insights 2021, 15, 11779322211025876. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.; Fleishon, S.; Kustin, T.; Dotan, E.; Mandelboim, M.; Erster, O.; Mendelson, E.; Mor, O.; Zuckerman, N.S.; Bucris, D.; et al. The Unique Evolutionary Dynamics of the SARS-CoV-2 Delta Variant-2 Sequencing. medRxiv 2021. [Google Scholar] [CrossRef]
- Buckley, P.R.; Lee, C.H.; Pereira Pinho, M.; Ottakandathil Babu, R.; Woo, J.; Antanaviciute, A.; Simmons, A.; Ogg, G. HLA-Dependent Variation in SARS-CoV-2 CD8+ T Cell Cross-Reactivity with Human Coronaviruses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, S.; Ren, L.; Zheng, P.; Hu, X.; Jin, T.; Tan, X. Profiling CD8+ T Cell Epitopes of COVID-19 Convalescents Reveals Reduced Cellular Immune Responses to SARS-CoV-2 Variants. Cell Rep. 2021, 36, 109708. [Google Scholar] [CrossRef]
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and Cross-Reactive SARS-CoV-2 T Cell Epitopes in Unexposed Humans. Science 2020, 370, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-Reactive T Cells in Healthy Donors and Patients with COVID-19. Nature 2020, 587, 270–274. [Google Scholar] [CrossRef] [PubMed]
- le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-Specific T Cell Immunity in Cases of COVID-19 and SARS, and Uninfected Controls. Nature 2020, 584, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.K.; Hersby, D.S.; Tamhane, T.; Povlsen, H.R.; Amaya Hernandez, S.P.; Nielsen, M.; Gang, A.O.; Hadrup, S.R. SARS-CoV-2 Genome-Wide T Cell Epitope Mapping Reveals Immunodominance and Substantial CD8+ T Cell Activation in COVID-19 Patients. Sci. Immunol. 2021, 6, eabf7550. [Google Scholar] [CrossRef]
- Karami Fath, M.; Jahangiri, A.; Ganji, M.; Sefid, F.; Payandeh, Z.; Hashemi, Z.S.; Pourzardosht, N.; Hessami, A.; Mard-Soltani, M.; Zakeri, A.; et al. SARS-CoV-2 Proteome Harbors Peptides Which Are Able to Trigger Autoimmunity Responses: Implications for Infection, Vaccination, and Population Coverage. Front. Immunol. 2021, 12, 705772. [Google Scholar] [CrossRef] [PubMed]
- Balz, K.; Kaushik, A.; Chen, M.; Cemic, F.; Heger, V.; Renz, H.; Nadeau, K.; Skevaki, C. Homologies between SARS-CoV-2 and Allergen Proteins May Direct T Cell-Mediated Heterologous Immune Responses. Sci. Rep. 2021, 11, 4792–4798. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Sharmin, S.; Hong, J.; Lee, H.S.; Kim, H.J.; Hong, S.T. T Cell Epitopes of SARS-CoV-2 Spike Protein and Conserved Surface Protein of Plasmodium Malariae Share Sequence Homology. Open Life Sci. 2021, 16, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Haddad-Boubaker, S.; Othman, H.; Touati, R.; Ayouni, K.; Lakhal, M.; ben Mustapha, I.; Ghedira, K.; Kharrat, M.; Triki, H. In Silico Comparative Study of SARS-CoV-2 Proteins and Antigenic Proteins in BCG, OPV, MMR and Other Vaccines: Evidence of a Possible Putative Protective Effect. BMC Bioinform. 2021, 22, 163–176. [Google Scholar] [CrossRef]
- Snyder, T.M.; Gittelman, R.M.; Klinger, M.; May, D.H.; Osborne, E.J.; Taniguchi, R.; Zahid, H.J.; Kaplan, I.M.; Dines, J.N.; Noakes, M.T.; et al. Magnitude and Dynamics of the T-Cell Response to SARS-CoV-2 Infection at Both Individual and Population Levels. medRxiv 2020. [Google Scholar] [CrossRef]
- Prachar, M.; Justesen, S.; Steen-Jensen, D.B.; Thorgrimsen, S.; Jurgons, E.; Winther, O.; Bagger, F.O. Identification and Validation of 174 COVID-19 Vaccine Candidate Epitopes Reveals Low Performance of Common Epitope Prediction Tools. Sci. Rep. 2020, 10, 20465. [Google Scholar] [CrossRef]
- Ibarrondo, F.J.; Fulcher, J.A.; Goodman-Meza, D.; Elliott, J.; Hofmann, C.; Hausner, M.A.; Ferbas, K.G.; Tobin, N.H.; Aldrovandi, G.M.; Yang, O.O. Rapid Decay of Anti–SARS-CoV-2 Antibodies in Persons with Mild COVID-19. N. Engl. J. Med. 2020, 383, 1085–1087. [Google Scholar] [CrossRef]
- Kreer, C.; Zehner, M.; Weber, T.; Ercanoglu, M.S.; Gieselmann, L.; Rohde, C.; Halwe, S.; Korenkov, M.; Schommers, P.; Vanshylla, K.; et al. Longitudinal Isolation of Potent Near-Germline SARS-CoV-2-Neutralizing Antibodies from COVID-19 Patients. Cell 2020, 182, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.X.; Liu, B.Z.; Deng, H.J.; Wu, G.C.; Deng, K.; Chen, Y.K.; Liao, P.; Qiu, J.F.; Lin, Y.; Cai, X.F.; et al. Antibody Responses to SARS-CoV-2 in Patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef]
- Ripperger, T.J.; Uhrlaub, J.L.; Watanabe, M.; Wong, R.; Castaneda, Y.; Pizzato, H.A.; Thompson, M.R.; Bradshaw, C.; Weinkauf, C.C.; Bime, C.; et al. Orthogonal SARS-CoV-2 Serological Assays Enable Surveillance of Low-Prevalence Communities and Reveal Durable Humoral Immunity. Immunity 2020, 53, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Chia, W.N.; Zhu, F.; Ong, S.W.X.; Young, B.E.; Fong, S.-W.; le Bert, N.; Tan, C.W.; Tiu, C.; Zhang, J.; Tan, S.Y.; et al. Dynamics of SARS-CoV-2 Neutralising Antibody Responses and Duration of Immunity: A Longitudinal Study. Lancet Microbe 2021, 2, e240–e249. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Jung, J.H.; Rha, M.-S.; Sa, M.; Choi, H.K.; Jeon, J.H.; Seok, H.; Park, D.W.; Park, S.-H.; Jeong, H.W.; Choi, W.S.; et al. SARS-CoV-2-Specific T Cell Memory Is Sustained in COVID-19 Convalescent Patients for 10 Months with Successful Development of Stem Cell-like Memory T Cells. Nat. Commun. 2021, 12, 4043. [Google Scholar] [CrossRef] [PubMed]
- Shanehbandi, D.; Majidi, J.; Kazemi, T.; Baradaran, B.; Aghebati-Maleki, L. CD20-Based Immunotherapy of B-Cell Derived Hematologic Malignancies. Curr. Cancer Drug Targets 2017, 17, 423–444. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Bange, E.M.; Han, N.A.; Wileyto, P.; Kim, J.Y.; Gouma, S.; Robinson, J.; Greenplate, A.R.; Hwee, M.A.; Porterfield, F.; Owoyemi, O.; et al. CD8+ T Cells Contribute to Survival in Patients with COVID-19 and Hematologic Cancer. Nat. Med. 2021, 27, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012. [Google Scholar] [CrossRef]
- Mallajosyula, V.; Ganjavi, C.; Chakraborty, S.; McSween, A.M.; Pavlovitch-Bedzyk, A.J.; Wilhelmy, J.; Nau, A.; Manohar, M.; Nadeau, K.C.; Davis, M.M. CD8+ T Cells Specific for Conserved Coronavirus Epitopes Correlate with Milder Disease in Patients with COVID-19. Sci. Immunol. 2021, 6, eabg5669. [Google Scholar] [CrossRef]
- Noh, J.Y.; Jeong, H.W.; Kim, J.H.; Shin, E.C. T Cell-Oriented Strategies for Controlling the COVID-19 Pandemic. Nat. Rev. Immunol. 2021, 21, 687–688. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fett, C.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Virus-Specific Memory CD8 T Cells Provide Substantial Protection from Lethal Severe Acute Respiratory Syndrome Coronavirus Infection. J. Virol. 2014, 88, 11034–11044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhao, J.; Mangalam, A.K.; Channappanavar, R.; Fett, C.; Meyerholz, D.K.; Agnihothram, S.; Baric, R.S.; David, C.S.; Perlman, S. Airway Memory CD4 + T Cells Mediate Protective Immunity against Emerging Respiratory Coronaviruses. Immunity 2016, 44, 1379–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, O.-W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.-J. Memory T Cell Responses Targeting the SARS Coronavirus Persist up to 11 Years Post-Infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef]
- Tumer, G.; Simpson, B.; Roberts, T.K. Genetics, Human Major Histocompatibility Complex (MHC). Available online: https://www.ncbi.nlm.nih.gov/books/NBK538218/ (accessed on 26 October 2021).
- Requena, D.; Médico, A.; Chacón, R.D.; Ramírez, M.; Marín-Sánchez, O. Identification of Novel Candidate Epitopes on SARS-CoV-2 Proteins for South America: A Review of HLA Frequencies by Country. Front. Immunol. 2020, 11, 2008–2023. [Google Scholar] [CrossRef]
- Sarma, V.R.; Olotu, F.A.; Soliman, M.E.S. Integrative Immunoinformatics Paradigm for Predicting Potential B-Cell and T-Cell Epitopes as Viable Candidates for Subunit Vaccine Design against COVID-19 Virulence. Biomed. J. 2021, 44, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Murdocca, M.; Citro, G.; Romeo, I.; Lupia, A.; Miersch, S.; Amadio, B.; Bonomo, A.; Rossi, A.; Sidhu, S.S.; Pandolfi, P.P.; et al. Peptide Platform as a Powerful Tool in the Fight against COVID-19. Viruses 2021, 13, 1667. [Google Scholar] [CrossRef]
- Susithra Priyadarshni, M.; Isaac Kirubakaran, S.; Harish, M.C. In Silico Approach to Design a Multi-Epitopic Vaccine Candidate Targeting the Non-Mutational Immunogenic Regions in Envelope Protein and Surface Glycoprotein of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 1–16. [Google Scholar] [CrossRef]
- Chukwudozie, O.S.; Gray, C.M.; Fagbayi, T.A.; Chukwuanukwu, R.C.; Oyebanji, V.O.; Bankole, T.T.; Adewole, R.A.; Daniel, E.M. Immuno-Informatics Design of a Multimeric Epitope Peptide Based Vaccine Targeting SARS-CoV-2 Spike Glycoprotein. PLoS ONE 2021, 16, e0248061. [Google Scholar] [CrossRef]
- Khan, M.T.; Islam, M.J.; Parihar, A.; Islam, R.; Jerin, T.J.; Dhote, R.; Ali, M.A.; Laura, F.K.; Halim, M.A. Immunoinformatics and Molecular Modeling Approach to Design Universal Multi-Epitope Vaccine for SARS-CoV-2. Inform. Med. Unlocked 2021, 24, 100578. [Google Scholar] [CrossRef]
- Rakib, A.; Sami, S.A.; Islam, M.A.; Ahmed, S.; Faiz, F.B.; Khanam, B.H.; Marma, K.K.S.; Rahman, M.; Uddin, M.M.N.; Nainu, F.; et al. Epitope-Based Immunoinformatics Approach on Nucleocapsid Protein of Severe Acute Respiratory Syndrome-Coronavirus-2. Molecules 2020, 25, 5088. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Sharma, G.; Lee, S.-S. Immunoinformatics Approach for the Identification and Characterization of T Cell and B Cell Epitopes towards the Peptide-Based Vaccine against SARS-CoV-2. Arch. Med. Res. 2021, 52, 362–370. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Mallick, B.; Sharma, G.; Lee, S.-S.; Chakraborty, C. Immunoinformatics Approach to Understand Molecular Interaction between Multi-Epitopic Regions of SARS-CoV-2 Spike-Protein with TLR4/MD-2 Complex. Infect. Genet. Evol. 2020, 85, 104587. [Google Scholar] [CrossRef]
- Jakhar, R.; Gakhar, S.K. An Immunoinformatics Study to Predict Epitopes in the Envelope Protein of SARS-CoV-2. Can. J. Infect. Dis. Med. Microbiol. 2020, 2020, 7079356. [Google Scholar] [CrossRef]
- Qiao, L.; Chen, M.; Li, S.; Hu, J.; Gong, C.; Zhang, Z.; Cao, X. A Peptide-Based Subunit Candidate Vaccine against SARS-CoV-2 Delivered by Biodegradable Mesoporous Silica Nanoparticles Induced High Humoral and Cellular Immunity in Mice. Biomater. Sci. 2021, 9, 7287–7296. [Google Scholar] [CrossRef]
- Rahman, N.; Ali, F.; Basharat, Z.; Shehroz, M.; Khan, M.K.; Jeandet, P.; Nepovimova, E.; Kuca, K.; Khan, H. Vaccine Design from the Ensemble of Surface Glycoprotein Epitopes of SARS-CoV-2: An Immunoinformatics Approach. Vaccines 2020, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Oladipo, E.K.; Ajayi, A.F.; Onile, O.S.; Ariyo, O.E.; Jimah, E.M.; Ezediuno, L.O.; Adebayo, O.I.; Adebayo, E.T.; Odeyemi, A.N.; Oyeleke, M.O.; et al. Designing a Conserved Peptide-Based Subunit Vaccine against SARS-CoV-2 Using Immunoinformatics Approach. Silico Pharmacol. 2021, 9, 8–28. [Google Scholar] [CrossRef]
- Waqas, M.; Haider, A.; Rehman, A.; Qasim, M.; Umar, A.; Sufyan, M.; Akram, H.N.; Mir, A.; Razzaq, R.; Rasool, D.; et al. Immunoinformatics and Molecular Docking Studies Predicted Potential Multiepitope-Based Peptide Vaccine and Novel Compounds against Novel SARS-CoV-2 through Virtual Screening. BioMed Res. Int. 2021, 2021, 1596834. [Google Scholar] [CrossRef]
- Al Saba, A.; Adiba, M.; Saha, P.; Hosen, M.I.; Chakraborty, S.; Nabi, A.H.M.N. An In-Depth in Silico and Immunoinformatics Approach for Designing a Potential Multi-Epitope Construct for the Effective Development of Vaccine to Combat against SARS-CoV-2 Encompassing Variants of Concern and Interest. Comput. Biol. Med. 2021, 136, 104703. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.N.; Ovsyannikova, I.G.; Kennedy, R.B.; Poland, G.A. Immunoinformatic Identification of B Cell and T Cell Epitopes in the SARS-CoV-2 Proteome. Sci. Rep. 2020, 10, 14179. [Google Scholar] [CrossRef]
- Gangaev, A.; Ketelaars, S.L.C.; Patiwael, S.; Dopler, A.; Hoefakker, K.; de Biasi, S.; Gibellini, L.; Mussini, C.; Guaraldi, G.; Girardis, M.; et al. Identification and characterization of a SARS-CoV-2 specific CD8+ T cell response with immunodominant features. Nat. Commun. 2021, 12, 2593–2606. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, A.P.; Kula, T.; Wang, Y.; Nguyen, D.M.V.; Weinheimer, A.; Dunlap, G.S.; Xu, Q.; Nabilsi, N.; Perullo, C.R.; Cristofaro, A.W.; et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 That Largely Reside Outside the Spike Protein. Immunity 2020, 53, 1095–1107. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.S.; Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. In Silico T Cell Epitope Identification for SARS-CoV-2: Progress and Perspectives. Adv. Drug Deliv. Rev. 2021, 171, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Sodsai, P.; Chia, A.; Moreau, E.; Chng, M.H.Y.; Tham, C.Y.L.; Ho, Z.Z.; Banu, N.; Hirankarn, N.; Bertoletti, A. Immunoprevalence and Immunodominance of HLA-Cw*0801-Restricted T Cell Response Targeting the Hepatitis B Virus Envelope Transmembrane Region. J. Virol. 2014, 88, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Variants | Number of Isolates |
|---|---|
| Alpha (B.1.1.7) | 158 |
| Beta (B.1.351) | 374 |
| Delta (B.1.617.2) | 1157 |
| Eta (B.1.525) | 436 |
| Gamma (P.1) | 9 |
| Iota (B.1.526) | 24 |
| Kappa (B.1.617.1) | 148 |
| Lambda (C.7) | 286 |
| Mu (B.1.621) | 18 |
| Protein | Size (aa) | Number of Immunogenic Epitopes | ||
|---|---|---|---|---|
| Reported in IEDB (T-Cell Assay Positive) | % Immunogenic Epitopes Per Protein | % Immunogenic Epitopes Per Total Reported in IEDB | ||
| ORF1ab | 7096 | 678 | 9.6 | 38.4 |
| Spike | 1273 | 578 | 4.5 | 32.7 |
| ORF3a | 275 | 88 | 32.0 | 5 |
| Envelope | 75 | 13 | 17.3 | 0.7 |
| Membrane | 222 | 131 | 59.0 | 7.4 |
| ORF6 | 61 | 18 | 29.5 | 1.0 |
| ORF7a | 121 | 28 | 23.1 | 1.6 |
| ORF7b | 43 | 3 | 7.0 | 0.2 |
| ORF8 | 121 | 37 | 30.6 | 2.1 |
| Nucleocapsid | 419 | 185 | 44.2 | 0.5 |
| ORF10 | 38 | 8 | 21.0 | 0.5 |
| Total epitopes | 1767 | |||
| HLA alleles | Populations | ORF1ab T-Cell Epitopes | SARS-CoV-2 T-Cell Epitopes | % ORF1ab/SARS-CoV-2 Epitopes in T-Cell Assay | ||||
|---|---|---|---|---|---|---|---|---|
| Total | T-Cell Assay | HLA Assay | Total | T-Cell Assay | HLA Assay | |||
| A*01:01 | GER | 54 | 48 | 12 | 96 | 85 | 21 | 56.47 |
| A*02:01 | GER INA | 138 | 82 | 86 | 224 | 156 | 126 | 52.56 |
| A*02:03 | INA THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| A*02:07 | THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| A*03:01 | GER | 42 | 17 | 33 | 69 | 37 | 45 | 45.95 |
| A*11:01 | GER INA THA | 49 | 19 | 39 | 69 | 33 | 48 | 57.58 |
| A*24:02 | GER INA THA | 64 | 45 | 29 | 129 | 100 | 47 | 45.00 |
| A*24:07 | INA THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| A*33:03 | INA THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| A*34:01 | INA | 0 | 0 | 0 | 0 | 0 | 0 | |
| B*07:02 | GER | 38 | 34 | 4 | 81 | 72 | 13 | 47.22 |
| B*08:01 | GER | 26 | 25 | 1 | 56 | 52 | 4 | 48.08 |
| B*13:01 | THA | 0 | 0 | 0 | 1 | 1 | 0 | 0.00 |
| B*15:01 | GER | 34 | 29 | 5 | 56 | 44 | 12 | 65.91 |
| B*15:02 | INA THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| B*15:13 | INA | 0 | 0 | 0 | 0 | 0 | 0 | |
| B*15:21 | INA | 0 | 0 | 0 | 0 | 0 | 0 | |
| B*18:01 | INA THA | 1 | 0 | 1 | 3 | 0 | 3 | |
| B*35:05 | INA | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | |
| B*38:02 | INA | 0 | 0 | 0 | 0 | 0 | 0 | |
| B*40:01 | GER THA | 41 | 18 | 28 | 67 | 33 | 41 | 54.55 |
| B*44:03 | INA THA | 11 | 11 | 0 | 25 | 25 | 0 | 44.00 |
| B*46:01 | THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| B*58:01 | INA THA | 6 | 0 | 6 | 14 | 0 | 14 | |
| DRB1*01:01 | GER | 8 | 4 | 7 | 61 | 8 | 59 | 50.00 |
| DRB1*03:01 | GER THA | 1 | 0 | 1 | 28 | 20 | 15 | 0.00 |
| DRB1*04:01 | GER | 24 | 2 | 24 | 156 | 5 | 156 | 40.00 |
| DRB1*04:05 | THA | 1 | 0 | 1 | 39 | 0 | 39 | |
| DRB1*07:01 | GER INA THA | 9 | 9 | 1 | 63 | 34 | 40 | 26.47 |
| DRB1*09:01 | THA | 1 | 0 | 1 | 38 | 0 | 38 | |
| DRB1*11:01 | GER INA | 1 | 0 | 1 | 42 | 12 | 32 | 0.00 |
| DRB1*12:02 | INA THA | 0 | 0 | 0 | 3 | 3 | 0 | 0.00 |
| DRB1*14:54 | THA | 0 | 0 | 0 | 0 | 0 | 0 | |
| DRB1*15:01 | GER INA THA | 17 | 14 | 7 | 83 | 50 | 58 | 28.00 |
| DRB1*15:02 | INA THA | 2 | 2 | 0 | 10 | 10 | 0 | 20.00 |
| DRB1*16:02 | INA THA | 0 | 0 | 0 | 8 | 8 | 0 | 0.00 |
| Start Residue | Peptide | HLA Class I Alleles | Immunogenicity Score |
|---|---|---|---|
| 295 | FMGRIRSVY | HLA-A*01:01, HLA-A*29:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:12, HLA-B*15:13, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*46:01 | 0.1259 |
| 541 | RVVRSIFSR | HLA-A*03:01, HLA-A*11:01, HLA-A*11:04, HLA-A*33:03, HLA-A*74:01 | 0.0318 |
| 611 | WLTNIFGTV | HLA-A*02:01, HLA-A*02:03 | 0.2972 |
| 806 | MVTNNTFTL | HLA-A*02:06, HLA-A*34:01, HLA-B*35:02, HLA-B*35:30, HLA-B*56:01, HLA-B*56:02, HLA-B*46:01 | 0.1578 |
| 899 | WSMATYYLF b | HLA-A*01:01, HLA-A*24:02, HLA-A*24:07, HLA-A*24:10, HLA-A*29:01, HLA-A*32:01, HLA-B*13:01, HLA-B*15:02, HLA-B*15:12, HLA-B*15:13, HLA-B*15:17, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*18:01, HLA-B*18:02, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*52:01, HLA-B*56:07, HLA-B*57:01, HLA-B*58:01, HLA-B*46:01 | 0.0071 |
| 1055 | VVVNAANVY a | HLA-A*26:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:12, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*35:01, HLA-B*46:01 | 0.1005 |
| 1140 | HEVLLAPLL c | HLA-B*13:01, HLA-B*18:01, HLA-B*18:02, HLA-B*37:01, HLA-B*38:02, HLA-B*40:01, HLA-B*40:02, HLA-B*40:06, HLA-B*41:01, HLA-B*44:03 | 0.0124 |
| 1247 | FLTENLLLY b | HLA-A*01:01, HLA-A*26:01, HLA-A*29:01 | 0.0808 |
| 1254 | LYIDINGNL | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.2138 |
| 1269 | LVSDIDITF a | HLA-B*15:02, HLA-B*15:13, HLA-B*15:17, HLA-B*15:21, HLA-B*35:01, HLA-B*35:02, HLA-B*35:05, HLA-B*35:30, HLA-B*57:01, HLA-B*58:01, HLA-B*46:01 | 0.2541 |
| 1366 | ILGTVSWNL b | HLA-A*02:01, HLA-A*02:07 | 0.1177 |
| 1674 | YLATALLTL a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07, HLA-B*46:01 | 0.0927 |
| 2175 | LLQLCTFTR | HLA-A*33:03, HLA-A*74:01 | 0.0568 |
| 2327 | FLAYILFTR | HLA-A*33:03, HLA-A*74:01 | 0.2496 |
| 2331 | ILFTRFFYV a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*74:01, HLA-B*08:01, HLA-A*02:07 | 0.3343 |
| 2350 | FSYFAVHFI | HLA-B*51:01, HLA-B*51:02, HLA-B*52:01 | 0.2893 |
| 2597 | FSSTFNVPM | HLA-B*15:10, HLA-B*15:21, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*56:02, HLA-B*46:01 | 0.1216 |
| 2629 | LSTFISAAR | HLA-A*33:03, HLA-A*34:01, HLA-A*74:01 | 0.1602 |
| 2784 | AIFYLITPV b,c | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*34:01, HLA-A*02:07 | 0.1750 |
| 2786 | FYLITPVHV a | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.2114 |
| 2787 | YLITPVHVM a | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*26:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:10, HLA-B*15:12, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*35:01, HLA-A*02:07, HLA-B*46:01 | 0.1617 |
| 2883 | FLPRVFSAV a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-B*08:01, HLA-A*02:07 | 0.0821 |
| 3059 | LAYYFMRFR a | HLA-A*33:03, HLA-A*74:01 | 0.0559 |
| 3060 | AYYFMRFRR | HLA-A*33:03, HLA-A*74:01 | 0.1234 |
| 3076 | VVAFNTLLF | HLA-A*24:07, HLA-A*29:01 | 0.1449 |
| 3121 | FLAHIQWMV a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07 | 0.1502 |
| 3137 | FWITIAYII d | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.3233 |
| 3152 | FYWFFSNYL | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.1404 |
| 3466 | VLAWLYAAV a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07 | 0.2772 |
| 3481 | FLNRFTTTL a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-B*08:01, HLA-A*02:07, HLA-B*46:01 | 0.2560 |
| 3582 | LLLTILTSL b,c | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-B*08:01, HLA-A*02:07 | 0.0907 |
| 3605 | LYENAFLPF | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.1584 |
| 3652 | VYMPASWVM a,b | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.0253 |
| 3684 | YASAVVLLI a,c | HLA-B*51:01, HLA-B*51:02, HLA-B*52:01, HLA-B*56:07, HLA-B*58:01 | 0.0489 |
| 3692 | ILMTARTVY a | HLA-A*29:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:12, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*35:05, HLA-B*35:30, HLA-B*46:01 | 0.1258 |
| 3752 | FLARGIVFM a,b,c | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07 | 0.3263 |
| 4030 | TMLFTMLRK b | HLA-A*03:01, HLA-A*11:01, HLA-A*11:04, HLA-A*74:01 | 0.0076 |
| 4265 | VLSFCAFAV b | HLA-A*02:01, HLA-A*02:07 | 0.1701 |
| 4513 | YTMADLVYA b | HLA-A*02:01, HLA-A*02:06, HLA-A*02:07 | 0.0262 |
| 4656 | YIKWDLLKY | HLA-A*01:01, HLA-A*26:01, HLA-A*29:01, HLA-B*15:02, HLA-B*15:12, HLA-B*15:21, HLA-B*46:01 | 0.0287 |
| 4698 | ILHCANFNV a | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07 | 0.0833 |
| 4723 | KIFVDGVPF | HLA-A*32:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:25, HLA-B*15:32 | 0.1614 |
| 4846 | YYRYNLPTM | HLA-A*24:02, HLA-A*24:10 | 0.0097 |
| 4862 | FVVEVVDKY a | HLA-A*26:01, HLA-A*29:01, HLA-A*34:01, HLA-B*15:21, HLA-B*35:01, HLA-B*35:30, HLA-B*46:01 | 0.0859 |
| 5024 | MASLVLARK a | HLA-A*03:01, HLA-A*11:01, HLA-A*11:04, HLA-A*30:01, HLA-A*33:03, HLA-A*34:01, HLA-A*74:01 | 0.0282 |
| 5132 | FVNEFYAYL a | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*26:01, HLA-A*34:01, HLA-A*02:07, HLA-B*46:01 | 0.2400 |
| 5245 | LMIERFVSL a | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*32:01, HLA-B*08:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:10, HLA-B*15:12, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*35:02, HLA-B*37:01, HLA-B*38:02, HLA-B*48:01, HLA-A*02:07, HLA-B*46:01 | 0.2427 |
| 5247 | IERFVSLAI | HLA-B*13:01, HLA-B*37:01, HLA-B*40:01, HLA-B*40:02, HLA-B*40:06, HLA-B*41:01, HLA-B*44:03, HLA-B*52:01 | 0.0326 |
| 5250 | FVSLAIDAY | HLA-A*01:01, HLA-A*26:01, HLA-A*29:01, HLA-A*34:01, HLA-B*15:02, HLA-B*15:21, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*46:01 | 0.1401 |
| 5273 | HLYLQYIRK b | HLA-A*03:01, HLA-A*11:01, HLA-A*11:04, HLA-A*74:01 | 0.0139 |
| 5614 | FAIGLALYY a,c | HLA-A*01:01, HLA-A*26:01, HLA-A*29:01, HLA-B*15:13, HLA-B*15:21, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*58:01, HLA-B*46:01 | 0.0918 |
| 5678 | YVFCTVNAL a | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*26:01, HLA-A*34:01, HLA-B*07:02, HLA-B*07:05, HLA-B*15:02, HLA-B*15:10, HLA-B*15:21, HLA-B*35:01, HLA-B*35:02, HLA-B*35:05, HLA-B*35:30, HLA-B*38:02, HLA-B*48:01, HLA-B*56:01, HLA-B*56:02, HLA-A*02:07, HLA-B*46:01 | 0.0778 |
| 6070 | FKHLIPLMY | HLA-A*29:01, HLA-B*18:02 | 0.0065 |
| 6108 | VLWAHGFEL a | HLA-A*02:01, HLA-A*02:06, HLA-A*02:07 | 0.3320 |
| 6506 | FELWAKRNI | HLA-B*40:01, HLA-B*40:02, HLA-B*40:06, HLA-B*41:01 | 0.0943 |
| 6585 | FRNARNGVL | HLA-B*15:10, HLA-B*27:06 | 0.1343 |
| 6700 | HLLIGLAKR | HLA-A*33:03, HLA-A*74:01 | 0.0599 |
| 6714 | FELEDFIPM b | HLA-B*13:01, HLA-B*15:10, HLA-B*18:01, HLA-B*18:02, HLA-B*37:01, HLA-B*38:02, HLA-B*40:01, HLA-B*40:02, HLA-B*40:06, HLA-B*41:01, HLA-B*44:03, HLA-B*48:01 | 0.3348 |
| 6748 | LLLDDFVEI a,b,c | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-B*52:01, HLA-A*02:07 | 0.2439 |
| 6848 | CQYLNTLTL | HLA-B*13:01, HLA-B*15:10, HLA-B*27:06, HLA-B*37:01, HLA-B*38:02, HLA-B*48:01, HLA-B*52:01 | 0.0312 |
| 6850 | YLNTLTLAV a,b | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07 | 0.0762 |
| 6885 | WLPTGTLLV | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*02:07 | 0.0892 |
| 6978 | YKLMGHFAW | HLA-B*18:01, HLA-B*18:02 | 0.0048 |
| 7019 | YVMHANYIF a | HLA-A*24:02, HLA-A*24:07, HLA-B*15:02, HLA-B*15:13, HLA-B*15:21, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*56:02, HLA-B*46:01 | 0.0822 |
| 7026 | IFWRNTNPI | HLA-A*24:02, HLA-A*24:07, HLA-A*24:10 | 0.1423 |
| Start Residue | Epitope Sequence | HLA DRB1 Alleles | IFNγ Score |
|---|---|---|---|
| 447 | NDNLLEILQKEKVNI | DRB1*12:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, | 0.1311 |
| 448 | DNLLEILQKEKVNIN | DRB1*12:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, | 0.0556 |
| 554 | TAQNSVRVLQKAAIT | DRB1*12:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, | 0.0684 |
| 736 | PKEIIFLEGETLPTE | DRB1*01:01, DRB1*12:02, DRB1*15:01, DRB1*15:02, DRB1*16:02, | 0.0771 |
| 1054 | PTVVVNAANVYLKHG | DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:54, DRB1*15:01, DRB1*15:02, DRB1*16:02 | 0.0917 |
| 1187 | VSSFLEMKSEKQVEQ | DRB1*04:01, DRB1*04:03, DRB1*04:05, DRB1*04:06, DRB1*10:01, | 0.0899 |
| 1211 | VKPFITESKPSVEQR | DRB1*08:03, DRB1*11:01, DRB1*13:02, DRB1*14:05, DRB1*14:07, | 0.3157 |
| 1349 | CKSAFYILPSIISNE | DRB1*01:01, DRB1*04:01, DRB1*04:05, DRB1*08:03, DRB1*10:01, DRB1*11:01, DRB1*15:02, DRB1*16:02, | 0.2898 |
| 1350 | KSAFYILPSIISNEK a | DRB1*01:01, DRB1*04:01, DRB1*04:03, DRB1*04:05, DRB1*04:06, DRB1*08:03, DRB1*10:01, DRB1*11:01, DRB1*12:02, DRB1*15:02, DRB1*16:02, | 0.3378 |
| 1355 | ILPSIISNEKQEILG | DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.4244 |
| 1356 | LPSIISNEKQEILGT | DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.3025 |
| 1357 | PSIISNEKQEILGTV | DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, | 0.5074 |
| 2944 | AYESLRPDTRYVLMD | DRB1*03:01, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, | 0.3078 |
| 2945 | YESLRPDTRYVLMDG | DRB1*03:01, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.1649 |
| 2958 | DGSIIQFPNTYLEGS | DRB1*04:02, DRB1*13:02, DRB1*15:01, DRB1*15:02, DRB1*16:02, | 0.2103 |
| 3815 | VSTQEFRYMNSQGLL | DRB1*01:01, DRB1*07:01, DRB1*09:01, DRB1*15:02, DRB1*16:02, | 0.0976 |
| 3944 | IASEFSSLPSYAAFA | DRB1*01:01, DRB1*04:01, DRB1*10:01, DRB1*15:02, DRB1*16:02, | 0.0754 |
| 3945 | ASEFSSLPSYAAFAT | DRB1*01:01, DRB1*04:01, DRB1*10:01, DRB1*15:02, DRB1*16:02, | 0.3973 |
| 3951 | LPSYAAFATAQEAYE | DRB1*04:01, DRB1*04:03, DRB1*04:05, DRB1*04:06, DRB1*08:03, | 0.0518 |
| 4457 | LIDSYFVVKRHTFSN | DRB1*08:03, DRB1*11:01, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:07, DRB1*14:54, | 0.1304 |
| 4458 | IDSYFVVKRHTFSNY | DRB1*08:03, DRB1*11:01, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:07, DRB1*14:54, | 0.1870 |
| 4560 | NPDILRVYANLGERV | DRB1*04:02, DRB1*08:03, DRB1*12:02, DRB1*15:01, DRB1*15:02, DRB1*16:02, | 0.2299 |
| 4561 | PDILRVYANLGERVR a | DRB1*04:02, DRB1*08:03, DRB1*13:02, DRB1*15:01, DRB1*15:02, DRB1*16:02, | 0.2616 |
| 4761 | KELLVYAADPAMHAA | DRB1*04:01, DRB1*04:02, DRB1*15:01, DRB1*15:02, DRB1*16:02, | 0.2258 |
| 4830 | KHFFFAQDGNAAISD | DRB1*01:01, DRB1*04:01, DRB1*10:01, DRB1*14:07, DRB1*16:02, | 0.4401 |
| 4933 | QMNLKYAISAKNRAR | DRB1*08:03, DRB1*10:01, DRB1*11:01, DRB1*13:02, DRB1*14:05, DRB1*14:07, | 0.4044 |
| 4934 | MNLKYAISAKNRART | DRB1*08:03, DRB1*10:01, DRB1*11:01, DRB1*13:02, DRB1*14:05, DRB1*14:07, | 0.4019 |
| 4935 | NLKYAISAKNRARTV | DRB1*08:03, DRB1*11:01, DRB1*13:02, DRB1*14:05, DRB1*14:07, | 0.5938 |
| 5019 | PNMLRIMASLVLARK a | DRB1*01:01, DRB1*12:02, DRB1*14:04, DRB1*15:01, DRB1*15:02, DRB1*16:02, | 0.3914 |
| 5717 | AKHYVYIGDPAQLPA | DRB1*04:01, DRB1*04:03, DRB1*04:05, DRB1*04:06, DRB1*08:03, DRB1*10:01, DRB1*16:02, | 0.1673 |
| 5775 | TVSALVYDNKLKAHK | DRB1*03:01, DRB1*11:01, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.3517 |
| 5776 | VSALVYDNKLKAHKD a | DRB1*03:01, DRB1*08:03, DRB1*11:01, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.2560 |
| 5777 | SALVYDNKLKAHKDK | DRB1*03:01, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.4910 |
| 5834 | VFISPYNSQNAVASK | DRB1*01:01, DRB1*04:01, DRB1*04:02, DRB1*10:01, DRB1*15:01, DRB1*15:02, | 0.2236 |
| 6046 | PTGYVDTPNNTDFSR | DRB1*04:01, DRB1*04:03, DRB1*04:05, DRB1*04:06, DRB1*08:03, | 0.0690 |
| 6454 | LENVAFNVVNKGHFD | DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.0787 |
| 6726 | TVKNYFITDAQTGSS | DRB1*01:01, DRB1*04:01, DRB1*07:01, DRB1*09:01, DRB1*10:01, DRB1*16:02, | 0.0871 |
| 7075 | KGRLIIRENNRVVIS | DRB1*04:02, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, DRB1*15:01, DRB1*15:02 | 0.7895 |
| 7076 | GRLIIRENNRVVISS | DRB1*04:02, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, DRB1*15:01, DRB1*15:02 | 0.7985 |
| 7077 | RLIIRENNRVVISSD | DRB1*13:02, DRB1*14:01, DRB1*14:05, DRB1*14:07, DRB1*14:54, | 0.8026 |
| Peptide | Allele | HLA | TAP | Cle | Comb | Aff(nM) | %Rank |
|---|---|---|---|---|---|---|---|
| FWITIAYII | HLA-A*24:02 | 0.604 | 0.566 | 0.463 | 0.722 | 250.54 | 0.8 |
| SWITIAYII | HLA-A*24:02 | 0.65 | 0.884 | 0.617 | 0.811 | 35.49 | 0.3 |
| FWITIAYII | HLA-A*24:07 | 0.497 | 0.566 | 0.463 | 0.615 | 220.88 | 0.8 |
| SWITIAYII | HLA-A*24:07 | 0.591 | 0.884 | 0.617 | 0.752 | 40.82 | 0.15 |
| FWITIAYII | HLA-A*24:10 | 0.8 | 0.566 | 0.463 | 0.918 | 61.69 | 0.8 |
| SWITIAYII | HLA-A*24:10 | 0.848 | 0.884 | 0.617 | 1.009 | 14.15 | 0.4 |
| Start | SARS-CoV-2 Peptide | Human Peptides | Human Proteins | HLA Allele Presenting the Human Peptide | IEDB Confirmation Assay |
|---|---|---|---|---|---|
| 1140 | HEVLLAPLL | AEVLLAPLL | HSVI binding protein (AAF76892.1) | HLA-B*37:01, HLA-B*38:02, HLA-B*40:01, HLA-B*40:02, HLA-B*40:06, HLA-B*41:01, HLA-B*44:03, HLA-B*13:01 | n.a. |
| 2784 | AIFYLITPV | AIFYLITLV | olfactory receptor, family 2, subfamily W, member 1, isoform CRA_b (EAX03180.1) | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06 | T-cell assay and HLA assay |
| 3582 | LLLTILTSL | LLLTILTRP | hCG2023968 (EAW49626.1) | non binder | HLA assay |
| 3684 | YASAVVLLI | VASAVVLLG | molybdenum cofactor biosynthesis protein 1 isoform 7 (NP_001345459.1) | non-binder | T-cell assay |
| 3752 | FLARGIVFM | XCARGIVFM | immunoglobulin heavy chain junction region (MOL38621.1) | cannot generate a similar peptide, since the sequence is at the N-terminal end of the protein. | T-cell assay and HLA assay |
| 5614 | FAIGLALYY | SYIGLALYY | immunoglobulin heavy chain junction region (MOJ91547.1) | HLA-A*29:01 | T cell assay |
| 6748 | LLLDDFVEI | IALDDFVEI | Wolfram syndrome 1 (wolframin), isoform CRA_a (EAW82396.1) | HLA-A*02:06, HLA-B*52:01 | T-cell assay and HLA assay |
| Start Residue | Peptide and Entropy * | HLA Alleles Bind to the Peptides | Population Coverage | |||
|---|---|---|---|---|---|---|
| Indonesia | Thailand | Germany | World | |||
| 899 | WSMATYYLF (0.083) | HLA-A*01:01, HLA-A*24:02, HLA-A*24:07, HLA-A*24:10, HLA-A*29:01, HLA-A*32:01, HLA-B*13:02, HLA-B*15:02, HLA-B*15:12, HLA-B*15:13, HLA-B*15:17, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*18:01, HLA-B*18:02, HLA-B*35:01, HLA-B*35:05, HLA-B*35:30, HLA-B*52:01, HLA-B*56:07, HLA-B*57:01, HLA-B*58:01, HLA-B*46:01 | 94.80 | 77.44; | 66.25; | 64.13 |
| 5678 | YVFCTVNAL (0.026) | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*26:01, HLA-A*34:01, HLA-B*07:02, HLA-B*07:05, HLA-B*15:02, HLA-B*15:10, HLA-B*15:21, HLA-B*35:01, HLA-B*35:02, HLA-B*35:05, HLA-B*35:30, HLA-B*38:02, HLA-B*48:01, HLA-B*56:01, HLA-B*56:02, HLA-A*02:07, HLA-B*46:01 | 77.39 | 75.05 | 72.07 | 65.66 |
| 5245 | LMIERFVSL (0.000) | HLA-A*02:01, HLA-A*02:03, HLA-A*02:06, HLA-A*32:01, HLA-B*08:01, HLA-B*15:01, HLA-B*15:02, HLA-B*15:10, HLA-B*15:12, HLA-B*15:21, HLA-B*15:25, HLA-B*15:32, HLA-B*35:02, HLA-B*37:01, HLA-B*38:02, HLA-B*48:01, HLA-A*02:07, HLA-B*46:01 | 63.65 | 74.26 | 71.42 | 63.19 |
| 6714 | FELEDFIPM (0.037) | HLA-B*13:01, HLA-B*13:02, HLA-B*15:10, HLA-B*18:01, HLA-B*18:02, HLA-B*37:01, HLA-B*38:02, HLA-B*40:01, HLA-B*40:02, HLA-B*40:06, HLA-B*41:01, HLA-B*44:03, HLA-B*48:01 | 51.64 | 46.46 | 35.87 | 35.59 |
| 5024 | MASLVLARK (0.000) | HLA-A*03:01, HLA-A*11:01, HLA-A*11:04, HLA-A*30:01, HLA-A*33:03, HLA-A*34:01, HLA-A*74:01 | 67.42 | 55.82 | 40.12 | 40.42 |
| 6848 | CQYLNTLTL (0.000) | HLA-B*13:02, HLA-B*15:10, HLA-B*27:06, HLA-B*37:01, HLA-B*38:02, HLA-B*48:01, HLA-B*52:01 | 21.20 | 20.90 | 10.15 | 13.16 |
| 2350 | FSYFAVHFI (0.027) | HLA-B*51:01, HLA-B*52:01 | 8.29 | 13.51 | 12.13 | 10.26 |
| 1350 | KSAFYILPSIISNEK (0.0283; 0.027; 0.023; 0.015; 0.015; 0.013; 0.013) | DRB1*01:01, DRB1*04:01, DRB1*04:03, DRB1*04:05, DRB1*04:06, DRB1*08:03, DRB1*10:01, DRB1*11:01, DRB1*12:02, DRB1*15:02, DRB1*16:02 | 91.13 | 74.99 | 47.87 | 47.60 |
| 7076 | GRLIIRENNRVVISS (0.000; 0.000; 0.000; 0.000; 0.000; 0.000; 0.000) | DRB1*04:02, DRB1*13:02, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54, DRB1*15:01, DRB1*15:02 | 53.28 | 50.97 | 40.57 | 37.72 |
| 7077 | RLIIRENNRVVISSD (0.000; 0.000; 0.000; 0.000; 0.000; 0.000; 0.000) | DRB1*13:02, DRB1*14:01, DRB1*14:05, DRB1*14:07, DRB1*14:54 | 4.18 | 11.27 | 13.43 | 16.78 |
| 2944 | AYESLRPDTRYVLMD (0.068; 0.045; 0.039; 0.0505; 0.027; 0.028; 0.026) | DRB1*03:01, DRB1*14:01, DRB1*14:04, DRB1*14:05, DRB1*14:54 | 10.59 | 23.69 | 25.46 | 27.58 |
| 3815 | VSTQEFRYMNSQGLL (0.000; 0.000; 0.000; 0.000; 0.000; 0.000; 0.129) | DRB1*01:01, DRB1*07:01, DRB1*09:01, DRB1*15:02, DRB1*16:02 | 63.57 | 63.45 | 41.63 | 38.08 |
| Epitope set | 100.00 | 100.00 | 99.98 | 99.88 | ||
| Population/Area | Class I | Class II | Class Combined | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Coverage a | Average_Hit b | pc90 c | Coverage a | Average_Hit b | pc90 c | Coverage a | Average_Hit b | pc90 c | |
| Germany | 99.75% | 3.89 | 2.54 | 93.25% | 1.81 | 1.09 | 99.98% | 5.7 | 4.03 |
| Indonesia | 100.0% | 5.66 | 4.1 | 99.68% | 2.68 | 1.46 | 100.0% | 8.35 | 6.19 |
| Thailand | 99.76% | 4.82 | 2.9 | 98.69% | 2.63 | 1.48 | 100.0% | 7.45 | 5.08 |
| World | 98.77% | 3.65 | 2.08 | 90.66% | 1.82 | 1.02 | 99.88% | 5.47 | 3.38 |
| Average | 99.57 | 4.5 | 2.9 | 95.57 | 2.23 | 1.26 | 99.97 | 6.74 | 4.67 |
| Standard deviation | 0.47 | 0.8 | 0.75 | 3.75 | 0.42 | 0.21 | 0.05 | 1.2 | 1.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustiananda, M.; Sulistyo, B.P.; Agustriawan, D.; Andarini, S. Immunoinformatics Analysis of SARS-CoV-2 ORF1ab Polyproteins to Identify Promiscuous and Highly Conserved T-Cell Epitopes to Formulate Vaccine for Indonesia and the World Population. Vaccines 2021, 9, 1459. https://doi.org/10.3390/vaccines9121459
Gustiananda M, Sulistyo BP, Agustriawan D, Andarini S. Immunoinformatics Analysis of SARS-CoV-2 ORF1ab Polyproteins to Identify Promiscuous and Highly Conserved T-Cell Epitopes to Formulate Vaccine for Indonesia and the World Population. Vaccines. 2021; 9(12):1459. https://doi.org/10.3390/vaccines9121459
Chicago/Turabian StyleGustiananda, Marsia, Bobby Prabowo Sulistyo, David Agustriawan, and Sita Andarini. 2021. "Immunoinformatics Analysis of SARS-CoV-2 ORF1ab Polyproteins to Identify Promiscuous and Highly Conserved T-Cell Epitopes to Formulate Vaccine for Indonesia and the World Population" Vaccines 9, no. 12: 1459. https://doi.org/10.3390/vaccines9121459
APA StyleGustiananda, M., Sulistyo, B. P., Agustriawan, D., & Andarini, S. (2021). Immunoinformatics Analysis of SARS-CoV-2 ORF1ab Polyproteins to Identify Promiscuous and Highly Conserved T-Cell Epitopes to Formulate Vaccine for Indonesia and the World Population. Vaccines, 9(12), 1459. https://doi.org/10.3390/vaccines9121459