The Stem Cell Revolution Revealing Protozoan Parasites’ Secrets and Paving the Way towards Vaccine Development


{kind=link}
{kind=link}
Abstract
:1. Introduction
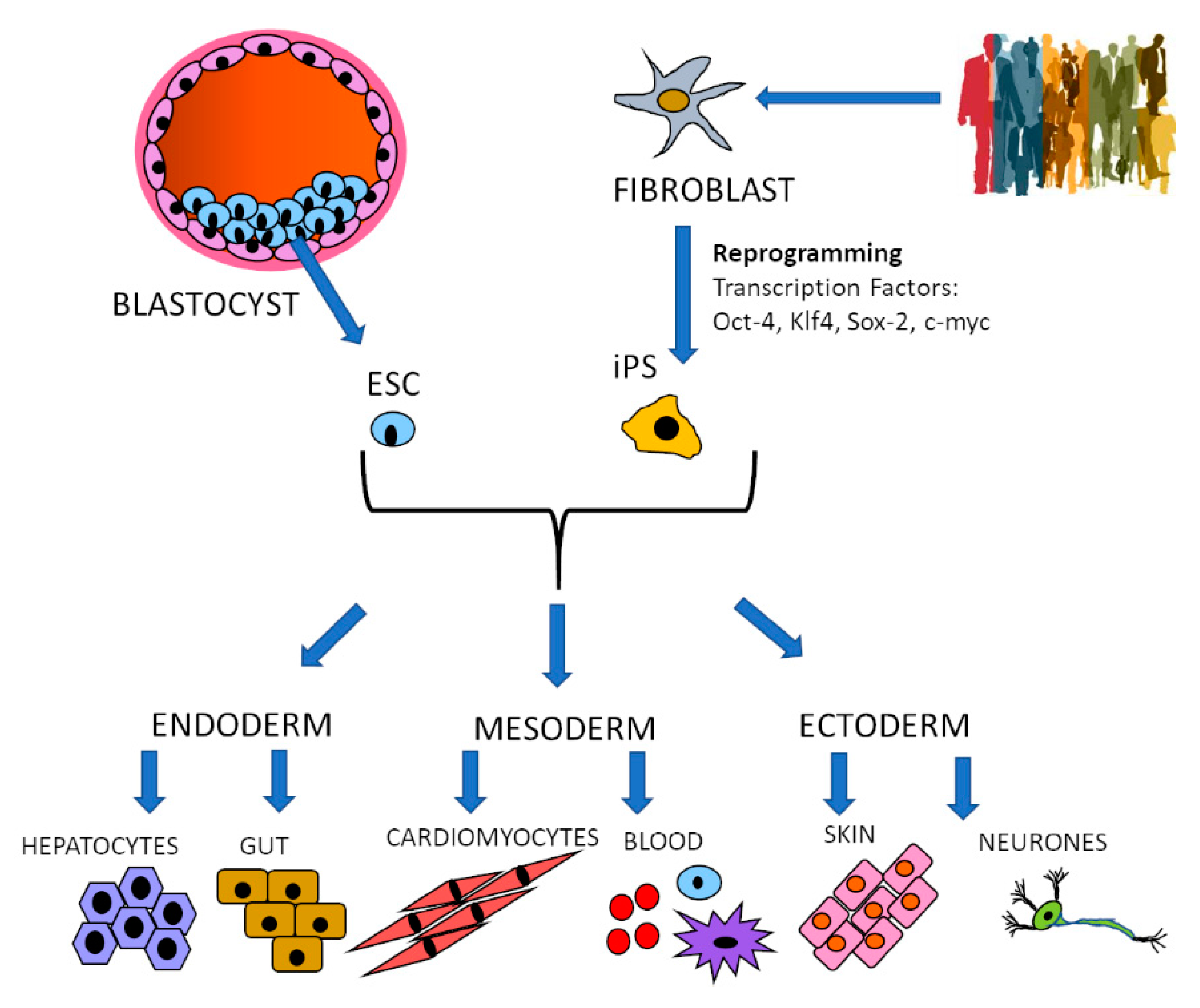
2. Specific Cell Type Generation: Stem Cells
2.1. Malaria
2.2. Chagas Disease
2.3. Leishmaniasis
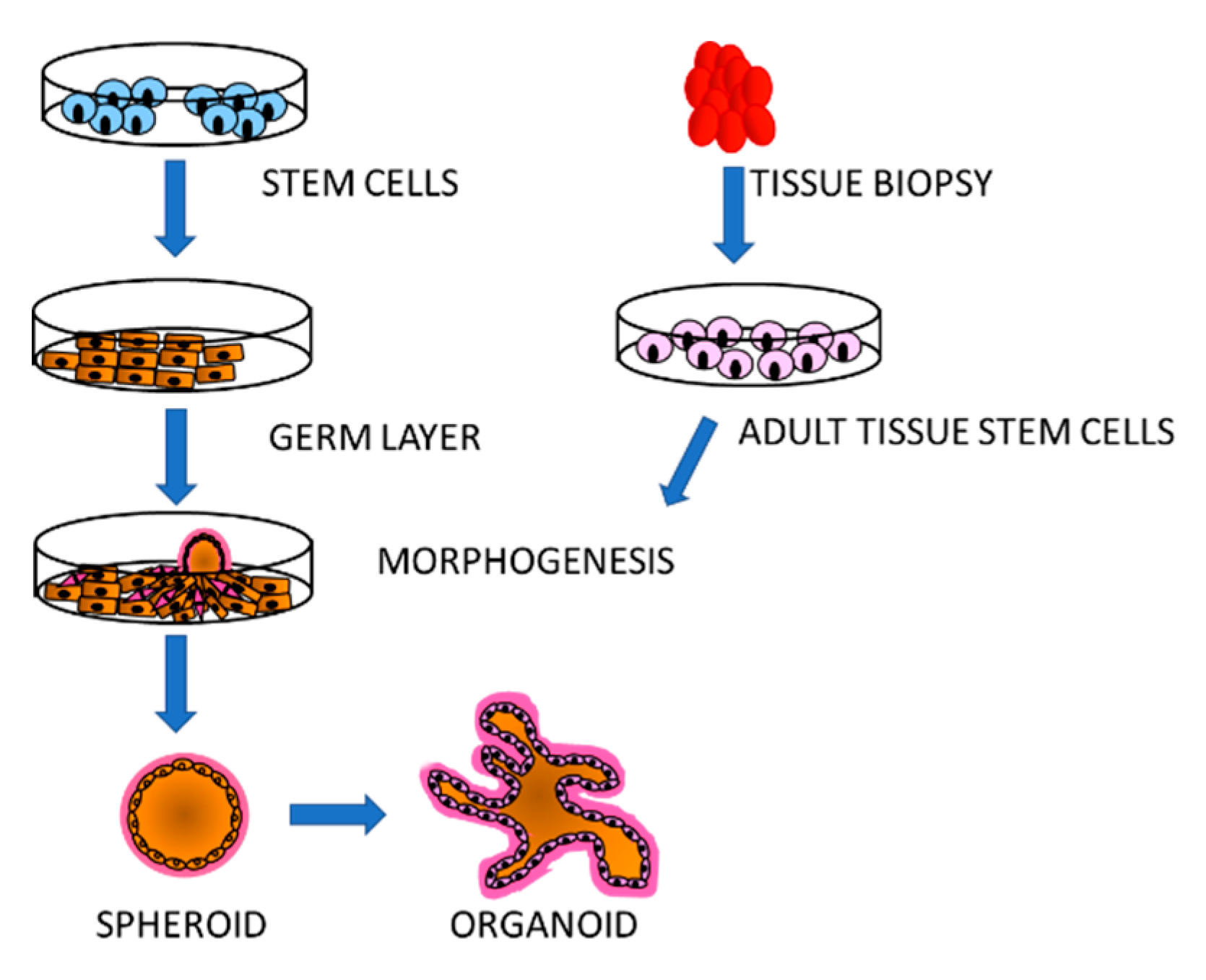
3. Simulation of Tissue Environment: Organoids
3.1. Malaria
3.2. Toxoplasmosis
3.3. Criptosporidiosis
4. Concluding Remarks and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grzybek, M.; Golonko, A.; Gorska, A.; Szczepaniak, K.; Strachecka, A.; Lass, A.; Lisowski, P. The CRISPR/Cas9 system sheds new lights on the biology of protozoan parasites. Appl. Microbiol. Biotechnol. 2018, 102, 4629–4640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, Y.R.; Sanchez, R. Update on Cyclospora cayetanensis, a food-borne and waterborne parasite. Clin. Microbiol. Rev. 2010, 23, 218–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunalan, K.; Rowley, E.H.; Miller, L.H. A Way Forward for Culturing Plasmodium vivax. Trends Parasitol. 2020, 36, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Arrowood, M.J. In vitro cultivation of cryptosporidium species. Clin. Microbiol. Rev. 2002, 15, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benere, E.; Geurden, T.; Robertson, L.; Van Assche, T.; Cos, P.; Maes, L. Infectivity of Giardia duodenalis Assemblages A and E for the gerbil and axenisation of duodenal trophozoites. Parasitol. Int. 2010, 59, 634–637. [Google Scholar] [CrossRef]
- Piras, R.; Piras, M.M.; Henriquez, D. Trypanosoma cruzi-fibroblastic cell interactions necessary for cellular invasion. Cytopathol. Parasit. Dis. 1983, 99, 31–51. [Google Scholar] [CrossRef]
- Klotz, C.; Aebischer, T.; Seeber, F. Stem cell-derived cell cultures and organoids for protozoan parasite propagation and studying host-parasite interaction. Int. J. Med. Microbiol. 2012, 302, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Yiangou, L.; Montandon, R.; Modrzynska, K.; Rosen, B.; Bushell, W.; Hale, C.; Billker, O.; Rayner, J.C.; Pance, A. A Stem Cell Strategy Identifies Glycophorin C as a Major Erythrocyte Receptor for the Rodent Malaria Parasite Plasmodium berghei. PLoS ONE 2016, 11, e0158238. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, D.A.; Betzer, D.P.; Smith, K.D.; Millman, Z.G.; Michalski, H.C.; Menchaca, S.E.; Zambriski, J.A.; Ojo, K.K.; Hulverson, M.A.; Arnold, S.L.; et al. Novel Bumped Kinase Inhibitors Are Safe and Effective Therapeutics in the Calf Clinical Model for Cryptosporidiosis. J. Infect. Dis. 2016, 214, 1856–1864. [Google Scholar] [CrossRef]
- Sheoran, A.; Wiffin, A.; Widmer, G.; Singh, P.; Tzipori, S. Infection with Cryptosporidium hominis provides incomplete protection of the host against Cryptosporidium parvum. J. Infect. Dis. 2012, 205, 1019–1023. [Google Scholar] [CrossRef] [Green Version]
- Klebanov, N. Genetic Predisposition to Infectious Disease. Cureus 2018, 10, e3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, K.W.; Howell, J.C.; Wells, J.M.; Spence, J.R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 2011, 6, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersema, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Vazin, T.; Freed, W.J. Human embryonic stem cells: Derivation, culture, and differentiation: A review. Restor. Neurol. Neurosci. 2010, 28, 589–603. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Elajami, M.K.; Daouk, R.; Jalloul, H.; Darwish, B.; Chalhoub, R.M.; Assi, S.; Chamaa, F.; Abou-Kheir, W. Stem Cells: In Sickness and in Health. Curr. Stem Cell Res. Ther. 2020. [Google Scholar] [CrossRef]
- Kumano, K.; Arai, S.; Kurokawa, M. Generation of iPS cells from normal and malignant hematopoietic cells. Int. J. Hematol. 2013, 98, 145–152. [Google Scholar] [CrossRef]
- Matsui, T.; Miyamoto, N.; Saito, F.; Shinozawa, T. Molecular Profiling of Human Induced Pluripotent Stem Cell-Derived Cells and their Application for Drug Safety Study. Curr. Pharm. Biotechnol. 2020, 21, 807–828. [Google Scholar] [CrossRef]
- World Health Organisation. World Malaria Report 2018. Available online: https://www.who.int/malaria/publications/world-malaria-report-2018/en/ (accessed on 10 December 2020).
- RTS,S Clinical Trials Partnership. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar] [CrossRef] [Green Version]
- Pance, A. Diversify and Conquer: The Vaccine Escapism of Plasmodium falciparum. Microorganisms 2020, 8, 1748. [Google Scholar] [CrossRef] [PubMed]
- Schafer, C.; Roobsoong, W.; Kangwanrangsan, N.; Bardelli, M.; Rawlinson, T.A.; Dambrauskas, N.; Trakhimets, O.; Parthiban, C.; Goswami, D.; Reynolds, L.M.; et al. A Humanized Mouse Model for Plasmodium vivax to Test Interventions that Block Liver Stage to Blood Stage Transition and Blood Stage Infection. iScience 2020, 23, 101381. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.K.; Tandel, N.; Deshpande, R.; Engelman, R.W.; Patel, S.D.; Tyagi, P. Humanized Mice Are Instrumental to the Study of Plasmodium falciparum Infection. Front. Immunol. 2018, 9, 2550. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, A.; Culleton, R. Non-human primate malaria parasites: Out of the forest and into the laboratory. Parasitology 2018, 145, 41–54. [Google Scholar] [CrossRef]
- Kudyba, H.M.; Cobb, D.W.; Florentin, A.; Krakowiak, M.; Muralidharan, V. CRISPR/Cas9 Gene Editing to Make Conditional Mutants of Human Malaria Parasite P. falciparum. J. Vis. Exp. 2018. [Google Scholar] [CrossRef]
- Mohring, F.; Hart, M.N.; Rawlinson, T.A.; Henrici, R.; Charleston, J.A.; Diez Benavente, E.; Patel, A.; Hall, J.; Almond, N.; Campino, S.; et al. Rapid and iterative genome editing in the malaria parasite Plasmodium knowlesi provides new tools for P. vivax research. eLife 2019, 8. [Google Scholar] [CrossRef]
- Egan, E.S.; Jiang, R.H.; Moechtar, M.A.; Barteneva, N.S.; Weekes, M.P.; Nobre, L.V.; Gygi, S.P.; Paulo, J.A.; Frantzreb, C.; Tani, Y.; et al. Malaria. A forward genetic screen identifies erythrocyte CD55 as essential for Plasmodium falciparum invasion. Science 2015, 348, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Bei, A.K.; Brugnara, C.; Duraisingh, M.T. In vitro genetic analysis of an erythrocyte determinant of malaria infection. J. Infect. Dis. 2010, 202, 1722–1727. [Google Scholar] [CrossRef] [Green Version]
- Crosnier, C.; Bustamante, L.Y.; Bartholdson, S.J.; Bei, A.K.; Theron, M.; Uchikawa, M.; Mboup, S.; Ndir, O.; Kwiatkowski, D.P.; Duraisingh, M.T.; et al. Basigin is a receptor essential for erythrocyte invasion by Plasmodium falciparum. Nature 2011, 480, 534–537. [Google Scholar] [CrossRef] [Green Version]
- Douglas, A.D.; Williams, A.R.; Illingworth, J.J.; Kamuyu, G.; Biswas, S.; Goodman, A.L.; Wyllie, D.H.; Crosnier, C.; Miura, K.; Wright, G.J.; et al. The blood-stage malaria antigen PfRH5 is susceptible to vaccine-inducible cross-strain neutralizing antibody. Nat. Commun. 2011, 2, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, R.O.; Silk, S.E.; Elias, S.C.; Miura, K.; Diouf, A.; Galaway, F.; de Graaf, H.; Brendish, N.J.; Poulton, I.D.; Griffiths, O.J.; et al. Human vaccination against RH5 induces neutralizing antimalarial antibodies that inhibit RH5 invasion complex interactions. JCI Insight 2017, 2, e96381. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, L.Y.; Bartholdson, S.J.; Crosnier, C.; Campos, M.G.; Wanaguru, M.; Nguon, C.; Kwiatkowski, D.P.; Wright, G.J.; Rayner, J.C. A full-length recombinant Plasmodium falciparum PfRH5 protein induces inhibitory antibodies that are effective across common PfRH5 genetic variants. Vaccine 2013, 31, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trakarnsanga, K.; Griffiths, R.E.; Wilson, M.C.; Blair, A.; Satchwell, T.J.; Meinders, M.; Cogan, N.; Kupzig, S.; Kurita, R.; Nakamura, Y.; et al. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 2017, 8, 14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satchwell, T.J.; Wright, K.E.; Haydn-Smith, K.L.; Sanchez-Roman Teran, F.; Moura, P.L.; Hawksworth, J.; Frayne, J.; Toye, A.M.; Baum, J. Genetic manipulation of cell line derived reticulocytes enables dissection of host malaria invasion requirements. Nat. Commun. 2019, 10, 3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, E.J.; Shabani, E.; Rangel, G.W.; Gruring, C.; Kanjee, U.; Clark, M.A.; Chaand, M.; Kurita, R.; Nakamura, Y.; Ferreira, M.U.; et al. Generation of an immortalized erythroid progenitor cell line from peripheral blood: A model system for the functional analysis of Plasmodium spp. invasion. Am. J. Hematol. 2019, 94, 963–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pance, A.L.B.; Mwikali, K.; Koutsourakis, M.; Agu, C.; Rouhani, F.; Ponstingl, H.; Montandon, R.; Law, F.; Rayner, J.C. Stem cells as a tool to study malaria: The host side of the infection. BioRxiv, in submission.
- Espinosa, D.A.; Vega-Rodriguez, J.; Flores-Garcia, Y.; Noe, A.R.; Munoz, C.; Coleman, R.; Bruck, T.; Haney, K.; Stevens, A.; Retallack, D.; et al. The Plasmodium falciparum Cell-Traversal Protein for Ookinetes and Sporozoites as a Candidate for Preerythrocytic and Transmission-Blocking Vaccines. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [Green Version]
- Ogwang, C.; Kimani, D.; Edwards, N.J.; Roberts, R.; Mwacharo, J.; Bowyer, G.; Bliss, C.; Hodgson, S.H.; Njuguna, P.; Viebig, N.K.; et al. Prime-boost vaccination with chimpanzee adenovirus and modified vaccinia Ankara encoding TRAP provides partial protection against Plasmodium falciparum infection in Kenyan adults. Sci. Transl. Med. 2015, 7, 286re285. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.; Schwartz, R.E.; March, S.; Galstian, A.; Gural, N.; Shan, J.; Prabhu, M.; Mota, M.M.; Bhatia, S.N. Human iPSC-derived hepatocyte-like cells support Plasmodium liver-stage infection in vitro. Stem Cell Rep. 2015, 4, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, J.; Segeritz, C.P.; Griffiths, G.; Bushell, W.; Vallier, L.; Skarnes, W.C.; Mota, M.M.; Billker, O. A Novel Chemically Differentiated Mouse Embryonic Stem Cell-Based Model to Study Liver Stages of Plasmodium berghei. Stem Cell Rep. 2020, 14, 1123–1134. [Google Scholar] [CrossRef]
- Breyner, N.M.; Hecht, M.; Nitz, N.; Rose, E.; Carvalho, J.L. In vitro models for investigation of the host-parasite interface-possible applications in acute Chagas disease. Acta Trop. 2020, 202, 105262. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, E.; Serna, C.; Fragoso, S.P.; Nagarkatti, R.; Debrabant, A. A novel nanoluciferase-based system to monitor Trypanosoma cruzi infection in mice by bioluminescence imaging. PLoS ONE 2018, 13, e0195879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.D.; Fortes Francisco, A.; Taylor, M.C.; Burrell-Saward, H.; McLatchie, A.P.; Miles, M.A.; Kelly, J.M. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cell. Microbiol. 2014, 16, 1285–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesley, M.; Moraes, A.; Rosa, A.C.; Lott Carvalho, J.; Shiroma, T.; Vital, T.; Dias, N.; de Carvalho, B.; do Amaral Rabello, D.; Borges, T.; et al. Correlation of Parasite Burden, kDNA Integration, Autoreactive Antibodies, and Cytokine Pattern in the Pathophysiology of Chagas Disease. Front. Microbiol. 2019, 10, 1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonseca-Berzal, C.; Aran, V.J.; Escario, J.A.; Gomez-Barrio, A. Experimental models in Chagas disease: A review of the methodologies applied for screening compounds against Trypanosoma cruzi. Parasitol. Res. 2018, 117, 3367–3380. [Google Scholar] [CrossRef]
- Cross, M.J.; Berridge, B.R.; Clements, P.J.; Cove-Smith, L.; Force, T.L.; Hoffmann, P.; Holbrook, M.; Lyon, A.R.; Mellor, H.R.; Norris, A.A.; et al. Physiological, pharmacological and toxicological considerations of drug-induced structural cardiac injury. Br. J. Pharmacol. 2015, 172, 957–974. [Google Scholar] [CrossRef]
- Da Silva Lara, L.; Andrade-Lima, L.; Magalhaes Calvet, C.; Borsoi, J.; Lopes Alberto Duque, T.; Henriques-Pons, A.; Souza Pereira, M.C.; Veiga Pereira, L. Trypanosoma cruzi infection of human induced pluripotent stem cell-derived cardiomyocytes: An in vitro model for drug screening for Chagas disease. Microbes Infect. 2018, 20, 312–316. [Google Scholar] [CrossRef]
- Sass, G.; Madigan, R.T.; Joubert, L.M.; Bozzi, A.; Sayed, N.; Wu, J.C.; Stevens, D.A. A Combination of Itraconazole and Amiodarone Is Highly Effective against Trypanosoma cruzi Infection of Human Stem Cell-Derived Cardiomyocytes. Am. J. Trop. Med. Hyg. 2019, 101, 383–391. [Google Scholar] [CrossRef]
- Sass, G.; Tsamo, A.T.; Chounda, G.A.M.; Nangmo, P.K.; Sayed, N.; Bozzi, A.; Wu, J.C.; Nkengfack, A.E.; Stevens, D.A. Vismione B Interferes with Trypanosoma cruzi Infection of Vero Cells and Human Stem Cell-Derived Cardiomyocytes. Am. J. Trop. Med. Hyg. 2019, 101, 1359–1368. [Google Scholar] [CrossRef]
- Bozzi, A.; Sayed, N.; Matsa, E.; Sass, G.; Neofytou, E.; Clemons, K.V.; Correa-Oliveira, R.; Stevens, D.A.; Wu, J.C. Using Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes as a Model to Study Trypanosoma cruzi Infection. Stem Cell Rep. 2019, 12, 1232–1241. [Google Scholar] [CrossRef] [Green Version]
- O’Keeffe, A.; Hale, C.; Cotton, J.A.; Yardley, V.; Gupta, K.; Ananthanarayanan, A.; Murdan, S.; Croft, S.L. Novel 2D and 3D Assays to Determine the Activity of Anti-Leishmanial Drugs. Microorganisms 2020, 8, 831. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Zampieri, R.A.; Shaw, J.J.; Floeter-Winter, L.M.; Laranjeira-Silva, M.F. One Health Approach to Leishmaniases: Understanding the Disease Dynamics through Diagnostic Tools. Pathogens 2020, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, J.F.; de Figueiredo, S.M.; Monteiro, F.M.; Rocha-Silva, F.; Gaciele-Melo, C.; Coelho, S.S.; Lyon, S.; Caligiorne, R.B. New Approaches on Leishmaniasis Treatment and Prevention: A Review of Recent Patents. Recent Pat. Endocr. Metab. Immune Drug Discov. 2015, 9, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Shankar, P.; Mishra, J.; Singh, S. Possibilities and challenges for developing a successful vaccine for leishmaniasis. Parasit Vectors 2016, 9, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattock, N.M.; Peters, W. The experimental chemotherapy of leishmaniasis. I: Techniques for the study of drug action in tissue culture. Ann. Trop. Med. Parasitol. 1975, 69, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Dermine, J.F.; Goyette, G.; Houde, M.; Turco, S.J.; Desjardins, M. Leishmania donovani lipophosphoglycan disrupts phagosome microdomains in J774 macrophages. Cell. Microbiol. 2005, 7, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Bosshart, H.; Heinzelmann, M. THP-1 cells as a model for human monocytes. Ann. Transl. Med. 2016, 4, 438. [Google Scholar] [CrossRef] [Green Version]
- Allahverdiyev, A.M.; Bagirova, M.; Elcicek, S.; Koc, R.C.; Baydar, S.Y.; Findikli, N.; Oztel, O.N. Adipose tissue-derived mesenchymal stem cells as a new host cell in latent leishmaniasis. Am. J. Trop. Med. Hyg. 2011, 85, 535–539. [Google Scholar] [CrossRef] [Green Version]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Izpisua Belmonte, J.C. Organoids-Preclinical Models of Human Disease. N. Engl. J. Med. 2019, 380, 569–579. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.A.; Min, S.; Kim, S.; Cho, S. Organoids for advanced therapeutics and disease models. Adv. Ther. 2019, 2, 1800087. [Google Scholar] [CrossRef] [Green Version]
- Harbuzariu, A.; Pitts, S.; Cespedes, J.C.; Harp, K.O.; Nti, A.; Shaw, A.P.; Liu, M.; Stiles, J.K. Modelling heme-mediated brain injury associated with cerebral malaria in human brain cortical organoids. Sci. Rep. 2019, 9, 19162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado Betancourt, E.; Hamid, B.; Fabian, B.T.; Klotz, C.; Hartmann, S.; Seeber, F. From Entry to Early Dissemination-Toxoplasma gondii’s Initial Encounter With Its Host. Front. Cell Infect. Microbiol. 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Flegr, J.; Prandota, J.; Sovickova, M.; Israili, Z.H. Toxoplasmosis--a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PLoS ONE 2014, 9, e90203. [Google Scholar] [CrossRef] [Green Version]
- Blader, I.J.; Coleman, B.I.; Chen, C.T.; Gubbels, M.J. Lytic Cycle of Toxoplasma gondii: 15 Years Later. Annu. Rev. Microbiol. 2015, 69, 463–485. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Grigg, M.E. Toxoplasma gondii: Laboratory Maintenance and Growth. Curr. Protoc. Microbiol. 2017, 44, 20C.1.1–20C.1.17. [Google Scholar] [CrossRef] [Green Version]
- Yui, S.; Nakamura, T.; Sato, T.; Nemoto, Y.; Mizutani, T.; Zheng, X.; Ichinose, S.; Nagaishi, T.; Okamoto, R.; Tsuchiya, K.; et al. Functional engraftment of colon epithelium expanded in vitro from a single adult Lgr5(+) stem cell. Nat. Med. 2012, 18, 618–623. [Google Scholar] [CrossRef]
- Hares, M.F.; Tiffney, E.A.; Johnston, L.J.; Luu, L.; Stewart, C.J.; Flynn, R.J.; Coombes, J.L. Stem cell-derived enteroid cultures as a tool for dissecting host-parasite interactions in the small intestinal epithelium. Parasite Immunol. 2020, e12765. [Google Scholar] [CrossRef]
- Luu, L.; Johnston, L.J.; Derricott, H.; Armstrong, S.D.; Randle, N.; Hartley, C.S.; Duckworth, C.A.; Campbell, B.J.; Wastling, J.M.; Coombes, J.L. An Open-Format Enteroid Culture System for Interrogation of Interactions Between Toxoplasma gondii and the Intestinal Epithelium. Front. Cell Infect. Microbiol. 2019, 9, 300. [Google Scholar] [CrossRef]
- Skorich, D.N.; Chiappino, M.L.; Nichols, B.A. Invasion of the guinea pig conjunctiva by Toxoplasma gondii. Investig. Ophthalmol. Vis. Sci. 1988, 29, 1871–1880. [Google Scholar]
- Tanaka, N.; Ashour, D.; Dratz, E.; Halonen, S. Use of human induced pluripotent stem cell-derived neurons as a model for Cerebral Toxoplasmosis. Microbes Infect. 2016, 18, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.H.; Han, H.W.; Lee, S.E.; Hong, S.H.; Cho, S.H.; Kim, S.C.; Koo, S.K.; Kim, J.H. Modelling Toxoplasma gondii infection in human cerebral organoids. Emerg. Microbes Infect. 2020, 9, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.; Zahedi, A.; O’Dea, M.; King, B.; Monis, P.; Thierry, B.; Carr, J.M.; Ryan, U. Organoids and Bioengineered Intestinal Models: Potential Solutions to the Cryptosporidium Culturing Dilemma. Microorganisms 2020, 8, 715. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.A.; Troeger, C.; Rao, P.C.; Blacker, B.F.; Brown, A.; Brewer, T.G.; Colombara, D.V.; De Hostos, E.L.; Engmann, C.; Guerrant, R.L.; et al. Morbidity, mortality, and long-term consequences associated with diarrhoea from Cryptosporidium infection in children younger than 5 years: A meta-analyses study. Lancet Glob. Health 2018, 6, e758–e768. [Google Scholar] [CrossRef] [Green Version]
- Mead, J.R. Prospects for immunotherapy and vaccines against Cryptosporidium. Hum. Vaccines Immunother. 2014, 10, 1505–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanis, P.; Aldeyarbi, H.M. Evolution of Cryptosporidium in vitro culture. Int. J. Parasitol. 2011, 41, 1231–1242. [Google Scholar] [CrossRef]
- Varughese, E.A.; Bennett-Stamper, C.L.; Wymer, L.J.; Yadav, J.S. A new in vitro model using small intestinal epithelial cells to enhance infection of Cryptosporidium parvum. J. Microbiol. Methods 2014, 106, 47–54. [Google Scholar] [CrossRef]
- Castellanos-Gonzalez, A.; Cabada, M.M.; Nichols, J.; Gomez, G.; White, A.C., Jr. Human primary intestinal epithelial cells as an improved in vitro model for Cryptosporidium parvum infection. Infect. Immun. 2013, 81, 1996–2001. [Google Scholar] [CrossRef] [Green Version]
- DeCicco RePass, M.A.; Chen, Y.; Lin, Y.; Zhou, W.; Kaplan, D.L.; Ward, H.D. Novel Bioengineered Three-Dimensional Human Intestinal Model for Long-Term Infection of Cryptosporidium parvum. Infect. Immun. 2017, 85, e00731-16. [Google Scholar] [CrossRef] [Green Version]
- Alcantara Warren, C.; Destura, R.V.; Sevilleja, J.E.; Barroso, L.F.; Carvalho, H.; Barrett, L.J.; O’Brien, A.D.; Guerrant, R.L. Detection of epithelial-cell injury, and quantification of infection, in the HCT-8 organoid model of cryptosporidiosis. J. Infect. Dis. 2008, 198, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, M.; Vanneste, S.B.; Creusy, C.; Guyot, K.; Gantois, N.; Chabe, M.; Delaire, B.; Mouray, A.; Baydoun, A.; Forzy, G.; et al. Three-dimensional (3D) culture of adult murine colon as an in vitro model of cryptosporidiosis: Proof of concept. Sci. Rep. 2017, 7, 17288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, I.; Dutta, D.; Schaefer, D.A.; Iakobachvili, N.; Artegiani, B.; Sachs, N.; Boonekamp, K.E.; Bowden, G.; Hendrickx, A.P.A.; Willems, R.J.L.; et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat. Microbiol. 2018, 3, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Heo, I.; O’Connor, R. Studying Cryptosporidium Infection in 3D Tissue-derived Human Organoid Culture Systems by Microinjection. J. Vis. Exp. 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.T.; Gong, A.Y.; Wang, Y.; Chen, X.; Lim, S.S.; Dolata, C.E.; Chen, X.M. Cryptosporidium parvum infection attenuates the ex vivo propagation of murine intestinal enteroids. Physiol. Rep. 2016, 4, e13060. [Google Scholar] [CrossRef]
- Wilke, G.; Funkhouser-Jones, L.J.; Wang, Y.; Ravindran, S.; Wang, Q.; Beatty, W.L.; Baldridge, M.T.; VanDussen, K.L.; Shen, B.; Kuhlenschmidt, M.S.; et al. A Stem-Cell-Derived Platform Enables Complete Cryptosporidium Development In Vitro and Genetic Tractability. Cell Host Microbe 2019, 26, 123–134.e128. [Google Scholar] [CrossRef] [Green Version]
- Wilke, G.; Wang, Y.; Ravindran, S.; Stappenbeck, T.; Witola, W.H.; Sibley, L.D. In Vitro Culture of Cryptosporidium parvum Using Stem Cell-Derived Intestinal Epithelial Monolayers. Methods Mol. Biol. 2020, 2052, 351–372. [Google Scholar] [CrossRef]
- Funkhouser-Jones, L.J.; Ravindran, S.; Sibley, L.D. Defining Stage-Specific Activity of Potent New Inhibitors of Cryptosporidium parvum Growth In Vitro. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Moreau, T.; Evans, A.L.; Vasquez, L.; Tijssen, M.R.; Yan, Y.; Trotter, M.W.; Howard, D.; Colzani, M.; Arumugam, M.; Wu, W.H.; et al. Large-scale production of megakaryocytes from human pluripotent stem cells by chemically defined forward programming. Nat. Commun. 2016, 7, 11208. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pance, A. The Stem Cell Revolution Revealing Protozoan Parasites’ Secrets and Paving the Way towards Vaccine Development. Vaccines 2021, 9, 105. https://doi.org/10.3390/vaccines9020105
Pance A. The Stem Cell Revolution Revealing Protozoan Parasites’ Secrets and Paving the Way towards Vaccine Development. Vaccines. 2021; 9(2):105. https://doi.org/10.3390/vaccines9020105
Chicago/Turabian StylePance, Alena. 2021. "The Stem Cell Revolution Revealing Protozoan Parasites’ Secrets and Paving the Way towards Vaccine Development" Vaccines 9, no. 2: 105. https://doi.org/10.3390/vaccines9020105
APA StylePance, A. (2021). The Stem Cell Revolution Revealing Protozoan Parasites’ Secrets and Paving the Way towards Vaccine Development. Vaccines, 9(2), 105. https://doi.org/10.3390/vaccines9020105