
HIV mRNA Vaccines—Progress and Future Paths


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Challenges of Developing an HIV Vaccine
2.1. Inducing Broadly Neutralizing Antibodies to HIV
2.2. Inducing CD8+ T Cell Responses to HIV through Vaccination
3. HIV mRNA Vaccine Platforms
3.1. Progress in mRNA Technology for HIV Vaccines
3.1.1. Non-Amplifying mRNA Vaccines
3.1.2. Self-Amplifying RNA Vaccines
3.1.3. Nucleoside Modification of mRNA
3.2. Progress in mRNA Delivery Strategies for HIV Vaccines
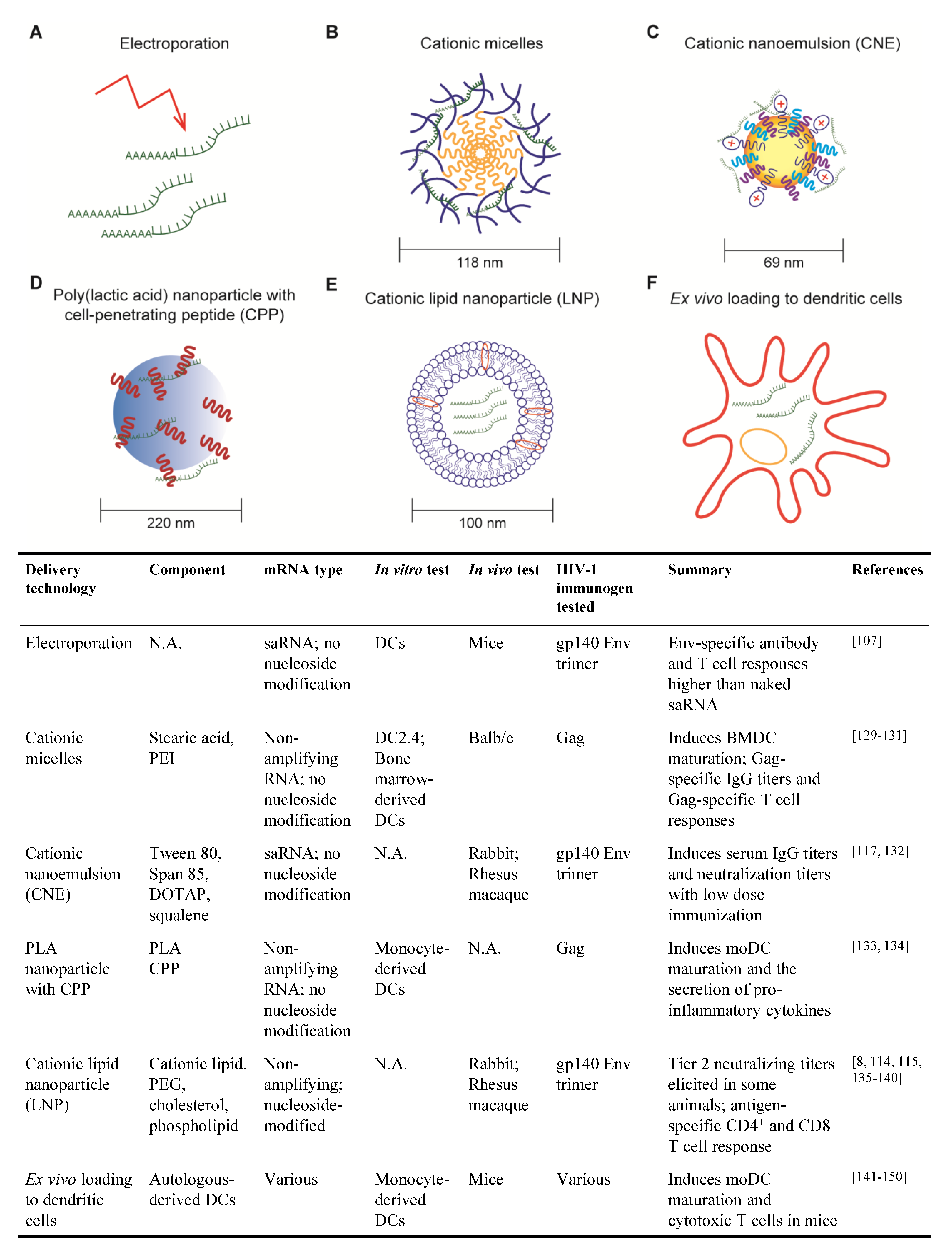
3.2.1. Electroporation
3.2.2. Cationic Micelles
3.2.3. Cationic Nanoemulsion
3.2.4. Poly (lactic acid) Nanoparticle with Cell-Penetrating Peptides
3.2.5. Cationic Lipid Nanoparticle
3.2.6. Ex Vivo Loading of Dendritic Cell
4. Overcoming the Challenges to HIV Vaccination with mRNA-Based Approaches
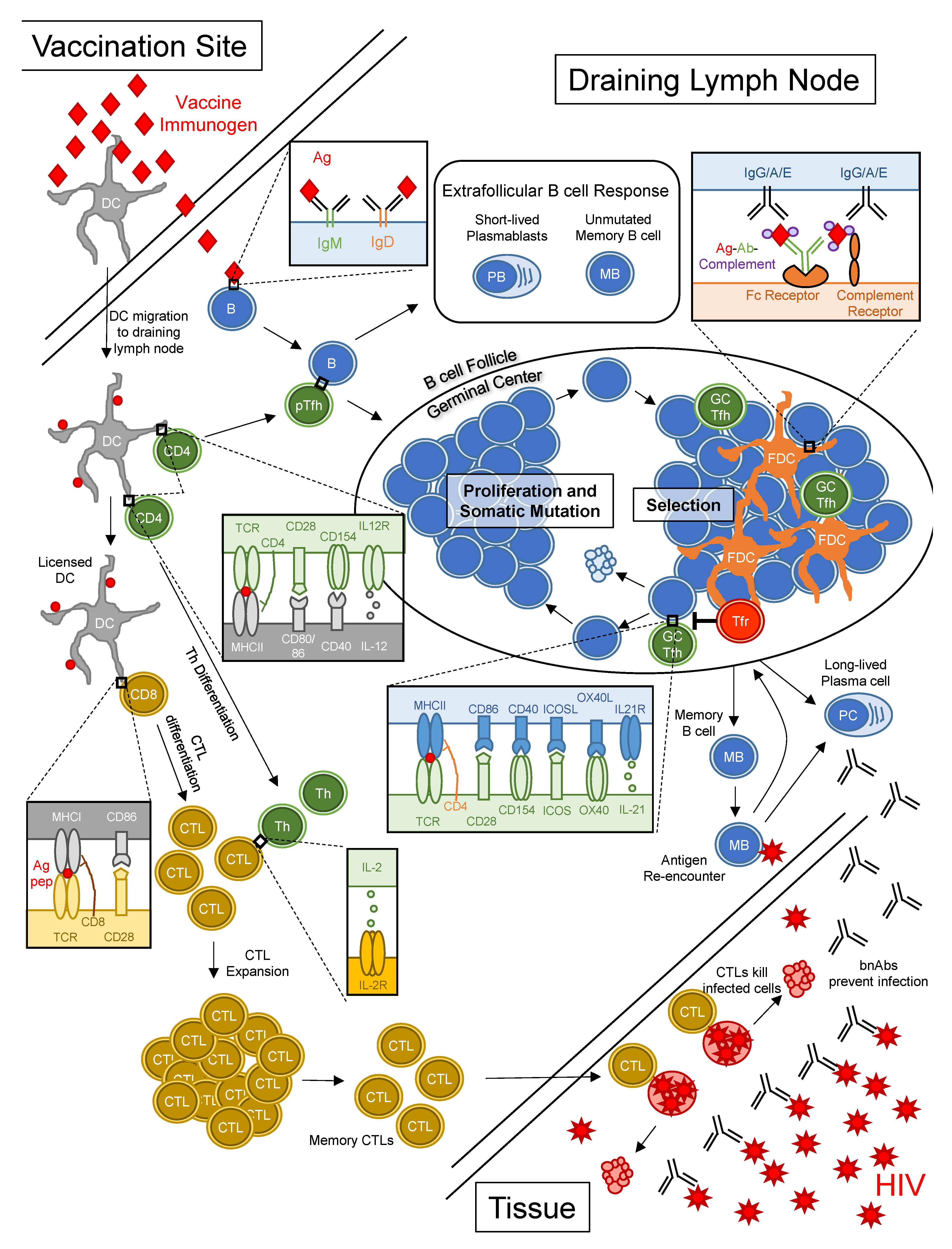
4.1. Challenge #1: B Cells with BCRs That Bind HIV Neutralizing Epitopes Are Rarely Generated and/or Are Auto-/Polyreactive and Thus Are Subject to Immune Tolerance
4.2. Challenge #2: bnAb Generation Requires High Levels of Improbable Mutations in Germinal Centers
4.3. Challenge #3: bnAb Induction by Vaccination will Require Multiple Immunogens for Sequential Immunizations
4.4. Challenge #4: bnAb and CD8+ T Cell Responses to HIV Are Optimally Induced via Different Pathways But Both May Be Required for Optimal Protection from HIV Infection
4.5. Unanswered Questions Surrounding mRNA Vaccines
5. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Barre-Sinoussi, F.; Ross, A.L.; Delfraissy, J.F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef]
- UNAIDS. UNAIDS Data. 2020. Available online: https://www.unaids.org/ (accessed on 30 November 2020).
- Gulick, R.M.; Flexner, C. Long-Acting HIV Drugs for Treatment and Prevention. Annu. Rev. Med. 2019, 70, 137–150. [Google Scholar] [CrossRef]
- Haynes, B.F.; Burton, D.R. HIV Developing an HIV vaccine What are the paths and obstacles to a practical vaccine? Science 2017, 355, 1129–1130. [Google Scholar] [CrossRef] [Green Version]
- Fruh, K.; Picker, L. CD8+ T cell programming by cytomegalovirus vectors: Applications in prophylactic and therapeutic vaccination. Curr. Opin. Immunol. 2017, 47, 52–56. [Google Scholar] [CrossRef]
- Jones, L.D.; Moody, M.A.; Thompson, A.B. Innovations in HIV-1 Vaccine Design. Clin. Ther. 2020, 42, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; DeKosky, B.J.; Ulmer, J.B. Antibody-guided structure-based vaccines. Semin. Immunol. 2020, 50, 101428. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef]
- Pardi, N.; Weissman, D. Nucleoside Modified mRNA Vaccines for Infectious Diseases. Methods Mol. Biol. 2017, 1499, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Kelsoe, G.; Harrison, S.C.; Kepler, T.B. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat. Biotechnol. 2012, 30, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.F.; Burton, D.R.; Mascola, J.R. Multiple roles for HIV broadly neutralizing antibodies. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arunachalam, P.S.; Charles, T.P.; Joag, V.; Bollimpelli, V.S.; Scott, M.K.D.; Wimmers, F.; Burton, S.L.; Labranche, C.C.; Petitdemange, C.; Gangadhara, S.; et al. T cell-inducing vaccine durably prevents mucosal SHIV infection even with lower neutralizing antibody titers. Nat. Med. 2020, 26, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Hangartner, L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu. Rev. Immunol. 2016, 34, 635–659. [Google Scholar] [CrossRef]
- Haynes, B.F.; Gilbert, P.B.; McElrath, M.J.; Zolla-Pazner, S.; Tomaras, G.D.; Alam, S.M.; Evans, D.T.; Montefiori, D.C.; Karnasuta, C.; Sutthent, R.; et al. Immune-Correlates Analysis of an HIV-1 Vaccine Efficacy Trial. N. Engl. J. Med. 2012, 366, 1275–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NIH. Experimental HIV Vaccine Regimen Ineffective in Preventing HIV. Available online: https://www.niaid.nih.gov/news-events/experimental-hiv-vaccine-regimen-ineffective-preventing-hiv (accessed on 5 January 2021).
- Hraber, P.; Seaman, M.S.; Bailer, R.T.; Mascola, J.R.; Montefiori, D.C.; Korber, B.T. Prevalence of broadly neutralizing antibody responses during chronic HIV-1 infection. AIDS 2014, 28, 163–169. [Google Scholar] [CrossRef]
- Escolano, A.; Dosenovic, P.; Nussenzweig, M.C. Progress toward active or passive HIV-1 vaccination. J. Exp. Med. 2017, 214, 3–16. [Google Scholar] [CrossRef]
- Burton, D.R.; Poignard, P.; Stanfield, R.L.; Wilson, I.A. Broadly Neutralizing Antibodies Present New Prospects to Counter Highly Antigenically Diverse Viruses. Science 2012, 337, 183–186. [Google Scholar] [CrossRef] [Green Version]
- Klein, F.; Mouquet, H.; Dosenovic, P.; Scheid, J.F.; Scharf, L.; Nussenzweig, M.C. Antibodies in HIV-1 Vaccine Development and Therapy. Science 2013, 341, 1199–1204. [Google Scholar] [CrossRef] [Green Version]
- Mascola, J.R.; Haynes, B.F. HIV-1 neutralizing antibodies: Understanding nature’s pathways. Immunol. Rev. 2013, 254, 225–244. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Martin, M.A. Of Mice, Macaques, and Men: Broadly Neutralizing Antibody Immunotherapy for HIV-1. Cell Host. Microbe. 2017, 22, 207–216. [Google Scholar] [CrossRef]
- Haynes, B.F.; Shaw, G.M.; Korber, B.; Kelsoe, G.; Sodroski, J.; Hahn, B.H.; Borrow, P.; McMichael, A.J. HIV-Host Interactions: Implications for Vaccine Design. Cell Host Microbe 2016, 19, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roark, R.S.; Li, H.; Williams, W.B.; Chug, H.; Mason, R.D.; Gorman, J.; Wang, S.; Lee, F.H.; Rando, J.; Bonsignori, M.; et al. Recapitulation of HIV-1 Env-antibody coevolution in macaques leading to neutralization breadth. Science 2020. [Google Scholar] [CrossRef]
- Doores, K.J.; Bonomelli, C.; Harvey, D.J.; Vasiljevic, S.; Dwek, R.A.; Burton, D.R.; Crispin, M.; Scanlan, C.N. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 13800–13805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Gaschen, B.; Yusim, K.; Thakallapally, R.; Kesmir, C.; Detours, V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 2001, 58, 19–42. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Decker, J.M.; Wang, S.; Hui, H.; Kappes, J.C.; Wu, X.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Kilby, J.M.; Saag, M.S.; et al. Antibody neutralization and escape by HIV-1. Nature 2003, 422, 307–312. [Google Scholar] [CrossRef]
- Richman, D.D.; Wrin, T.; Little, S.J.; Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 2003, 100, 4144–4149. [Google Scholar] [CrossRef] [Green Version]
- Seaman, M.S.; Janes, H.; Hawkins, N.; Grandpre, L.E.; Devoy, C.; Giri, A.; Coffey, R.T.; Harris, L.; Wood, B.; Daniels, M.G.; et al. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 2010, 84, 1439–1452. [Google Scholar] [CrossRef] [Green Version]
- Sarzotti-Kelsoe, M.; Bailer, R.T.; Turk, E.; Lin, C.L.; Bilska, M.; Greene, K.M.; Gao, H.; Todd, C.A.; Ozaki, D.A.; Seaman, M.S.; et al. Optimization and validation of the TZM-bl assay for standardized assessments of neutralizing antibodies against HIV-1. J. Immunol. Methods 2014, 409, 131–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montefiori, D.C.; Roederer, M.; Morris, L.; Seaman, M.S. Neutralization tiers of HIV-1. Curr. Opin. Hiv Aids 2018, 13, 128–136. [Google Scholar] [CrossRef]
- McCoy, L.E. The expanding array of HIV broadly neutralizing antibodies. Retrovirology 2018, 15, 70. [Google Scholar] [CrossRef]
- Shi, B.; Ma, L.; He, X.; Wang, X.; Wang, P.; Zhou, L.; Yao, X. Comparative analysis of human and mouse immunoglobulin variable heavy regions from IMGT/LIGM-DB with IMGT/HighV-QUEST. Theor. Biol. Med. Model 2014, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Sok, D.; Le, K.M.; Vadnais, M.; Saye-Francisco, K.L.; Jardine, J.G.; Torres, J.L.; Berndsen, Z.T.; Kong, L.; Stanfield, R.; Ruiz, J.; et al. Rapid elicitation of broadly neutralizing antibodies to HIV by immunization in cows. Nature 2017, 548, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Verkoczy, L.; Chen, Y.; Bouton-Verville, H.; Zhang, J.; Diaz, M.; Hutchinson, J.; Ouyang, Y.B.; Alam, S.M.; Holl, T.M.; Hwang, K.K.; et al. Rescue of HIV-1 broad neutralizing antibody-expressing B cells in 2F5 VH x VL knockin mice reveals multiple tolerance controls. J. Immunol. 2011, 187, 3785–3797. [Google Scholar] [CrossRef] [Green Version]
- Doyle-Cooper, C.; Hudson, K.E.; Cooper, A.B.; Ota, T.; Skog, P.; Dawson, P.E.; Zwick, M.B.; Schief, W.R.; Burton, D.R.; Nemazee, D. Immune tolerance negatively regulates B cells in knock-in mice expressing broadly neutralizing HIV antibody 4E10. J. Immunol. 2013, 191, 3186–3191. [Google Scholar] [CrossRef] [PubMed]
- Bonsignori, M.; Kreider, E.F.; Fera, D.; Meyerhoff, R.R.; Bradley, T.; Wiehe, K.; Alam, S.M.; Aussedat, B.; Walkowicz, W.E.; Hwang, K.K.; et al. Staged induction of HIV-1 glycan-dependent broadly neutralizing antibodies. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Kelsoe, G.; Haynes, B.F. Host controls of HIV broadly neutralizing antibody development. Immunol. Rev. 2017, 275, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sok, D.; Burton, D.R. Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol. 2018, 19, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Lynch, R.; Zhou, T.; Gao, F.; Alam, S.M.; Boyd, S.D.; Fire, A.Z.; Roskin, K.M.; Schramm, C.A.; Zhang, Z.; et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 2013, 496, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.L.; Gray, E.S.; Wibmer, C.K.; Bhiman, J.N.; Nonyane, M.; Sheward, D.J.; Hermanus, T.; Bajimaya, S.; Tumba, N.L.; Abrahams, M.R.; et al. Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat. Med. 2012, 18, 1688–1692. [Google Scholar] [CrossRef] [Green Version]
- Wibmer, C.K.; Bhiman, J.N.; Gray, E.S.; Tumba, N.; Abdool Karim, S.S.; Williamson, C.; Morris, L.; Moore, P.L. Viral escape from HIV-1 neutralizing antibodies drives increased plasma neutralization breadth through sequential recognition of multiple epitopes and immunotypes. PLoS Pathog. 2013, 9, e1003738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doria-Rose, N.A.; Schramm, C.A.; Gorman, J.; Moore, P.L.; Bhiman, J.N.; DeKosky, B.J.; Ernandes, M.J.; Georgiev, I.S.; Kim, H.J.; Pancera, M.; et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014, 509, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Chen, W.; Feng, Y.; Zhu, Z.; Prabakaran, P.; Wang, Y.; Zhang, M.Y.; Longo, N.S.; Dimitrov, D.S. Germline-like predecessors of broadly neutralizing antibodies lack measurable binding to HIV-1 envelope glycoproteins: Implications for evasion of immune responses and design of vaccine immunogens. Biochem. Biophys. Res. Commun. 2009, 390, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Kepler, T.B.; Munshaw, S.; Wiehe, K.; Zhang, R.; Yu, J.S.; Woods, C.W.; Denny, T.N.; Tomaras, G.D.; Alam, S.M.; Moody, M.A.; et al. Reconstructing a B-Cell Clonal Lineage. II. Mutation, Selection, and Affinity Maturation. Front. Immunol. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- LaBranche, C.C.; Henderson, R.; Hsu, A.; Behrens, S.; Chen, X.; Zhou, T.; Wiehe, K.; Saunders, K.O.; Alam, S.M.; Bonsignori, M.; et al. Neutralization-guided design of HIV-1 envelope trimers with high affinity for the unmutated common ancestor of CH235 lineage CD4bs broadly neutralizing antibodies. PLoS Pathog. 2019, 15, e1008026. [Google Scholar] [CrossRef] [Green Version]
- Bonsignori, M.; Scott, E.; Wiehe, K.; Easterhoff, D.; Alam, S.M.; Hwang, K.K.; Cooper, M.; Xia, S.M.; Zhang, R.; Montefiori, D.C.; et al. Inference of the HIV-1 VRC01 Antibody Lineage Unmutated Common Ancestor Reveals Alternative Pathways to Overcome a Key Glycan Barrier. Immunity 2018, 49, 1162–1174 e1168. [Google Scholar] [CrossRef]
- Jardine, J.; Julien, J.P.; Menis, S.; Ota, T.; Kalyuzhniy, O.; McGuire, A.; Sok, D.; Huang, P.S.; MacPherson, S.; Jones, M.; et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 2013, 340, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Jardine, J.G.; Ota, T.; Sok, D.; Pauthner, M.; Kulp, D.W.; Kalyuzhniy, O.; Skog, P.D.; Thinnes, T.C.; Bhullar, D.; Briney, B.; et al. HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 2015, 349, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardine, J.G.; Kulp, D.W.; Havenar-Daughton, C.; Sarkar, A.; Briney, B.; Sok, D.; Sesterhenn, F.; Ereno-Orbea, J.; Kalyuzhniy, O.; Deresa, I.; et al. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 2016, 351, 1458–1463. [Google Scholar] [CrossRef] [Green Version]
- Havenar-Daughton, C.; Sarkar, A.; Kulp, D.W.; Toy, L.; Hu, X.; Deresa, I.; Kalyuzhniy, O.; Kaushik, K.; Upadhyay, A.A.; Menis, S.; et al. The human naive B cell repertoire contains distinct subclasses for a germline-targeting HIV-1 vaccine immunogen. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Dosenovic, P.; von Boehmer, L.; Escolano, A.; Jardine, J.; Freund, N.T.; Gitlin, A.D.; McGuire, A.T.; Kulp, D.W.; Oliveira, T.; Scharf, L.; et al. Immunization for HIV-1 Broadly Neutralizing Antibodies in Human Ig Knockin Mice. Cell 2015, 161, 1505–1515. [Google Scholar] [CrossRef] [Green Version]
- McGuire, A.T.; Gray, M.D.; Dosenovic, P.; Gitlin, A.D.; Freund, N.T.; Petersen, J.; Correnti, C.; Johnsen, W.; Kegel, R.; Stuart, A.B.; et al. Specifically modified Env immunogens activate B-cell precursors of broadly neutralizing HIV-1 antibodies in transgenic mice. Nat. Commun. 2016, 7, 10618. [Google Scholar] [CrossRef]
- Medina-Ramirez, M.; Garces, F.; Escolano, A.; Skog, P.; de Taeye, S.W.; Del Moral-Sanchez, I.; McGuire, A.T.; Yasmeen, A.; Behrens, A.J.; Ozorowski, G.; et al. Design and crystal structure of a native-like HIV-1 envelope trimer that engages multiple broadly neutralizing antibody precursors in vivo. J. Exp. Med. 2017, 214, 2573–2590. [Google Scholar] [CrossRef]
- Saunders, K.O.; Wiehe, K.; Tian, M.; Acharya, P.; Bradley, T.; Alam, S.M.; Go, E.P.; Scearce, R.; Sutherland, L.; Henderson, R.; et al. Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science 2019, 366, 1215. [Google Scholar] [CrossRef] [PubMed]
- Wiehe, K.; Bradley, T.; Meyerhoff, R.R.; Hart, C.; Williams, W.B.; Easterhoff, D.; Faison, W.J.; Kepler, T.B.; Saunders, K.O.; Alam, S.M.; et al. Functional Relevance of Improbable Antibody Mutations for HIV Broadly Neutralizing Antibody Development. Cell Host. Microbe. 2018, 23, 759. [Google Scholar] [CrossRef] [Green Version]
- Victora, G.D.; Nussenzweig, M.C. Germinal centers. Annu. Rev. Immunol. 2012, 30, 429–457. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Kageyama, R.; Eto, D.; Escobar, T.C.; Johnston, R.J.; Monticelli, L.; Lao, C.; Crotty, S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011, 34, 932–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardtke, S.; Ohl, L.; Forster, R. Balanced expression of CXCR5 and CCR7 on follicular T helper cells determines their transient positioning to lymph node follicles and is essential for efficient B-cell help. Blood 2005, 106, 1924–1931. [Google Scholar] [CrossRef] [Green Version]
- Haynes, N.M.; Allen, C.D.C.; Lesley, R.; Ansel, K.M.; Killeen, N.; Cyster, J.G. Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1(High) germinal center-associated subpopulation. J. Immunol. 2007, 179, 5099–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Tew, J.G.; Mandel, T.E.; Rice, P.L. Immune Elimination and Immune Retention the Relationship between Antigen Retained in the Foot and the Elicitation of Footpad Swelling. Immunology 1980, 40, 425–433. [Google Scholar]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3(+) follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, U975–U995. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, I.; Agua-Doce, A.; Hernandez, A.; Almeida, C.; Oliveira, V.G.; Faro, J.; Graca, L. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J. Immunol. 2011, 187, 4553–4560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S. Follicular Helper CD4 T Cells (T-FH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef]
- Jerne, N.K. A study of avidity based on rabbit skin responses to diphtheria toxin-antitoxin mixtures. Acta Pathol. Microbiol. Scand. Suppl. 1951, 87, 1–183. [Google Scholar]
- Eisen, H.N.; Siskind, G.W. Variations in Affinities of Antibodies during the Immune Response. Biochemistry 1964, 3, 996–1008. [Google Scholar] [CrossRef]
- Andrabi, R.; Bhiman, J.N.; Burton, D.R. Strategies for a multi-stage neutralizing antibody-based HIV vaccine. Curr. Opin. Immunol. 2018, 53, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Koup, R.A.; Ho, D.D. Shutting down HIV. Nature 1994, 370, 416. [Google Scholar] [CrossRef]
- Borrow, P.; Lewicki, H.; Hahn, B.H.; Shaw, G.M.; Oldstone, M.B. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994, 68, 6103–6110. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; O’Connor, D.H.; Jing, P.; Dzuris, J.L.; Mothe, B.R.; Vogel, T.U.; Dunphy, E.; Liebl, M.E.; Emerson, C.; Wilson, N.; et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 2000, 407, 386–390. [Google Scholar] [CrossRef]
- O’Connor, D.H.; Allen, T.M.; Vogel, T.U.; Jing, P.; DeSouza, I.P.; Dodds, E.; Dunphy, E.J.; Melsaether, C.; Mothe, B.; Yamamoto, H.; et al. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat. Med. 2002, 8, 493–499. [Google Scholar] [CrossRef]
- Goonetilleke, N.; Liu, M.K.; Salazar-Gonzalez, J.F.; Ferrari, G.; Giorgi, E.; Ganusov, V.V.; Keele, B.F.; Learn, G.H.; Turnbull, E.L.; Salazar, M.G.; et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 2009, 206, 1253–1272. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef]
- Carrington, M.; O’Brien, S.J. The influence of HLA genotype on AIDS. Annu. Rev. Med. 2003, 54, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellay, J.; Shianna, K.V.; Ge, D.; Colombo, S.; Ledergerber, B.; Weale, M.; Zhang, K.; Gumbs, C.; Castagna, A.; Cossarizza, A.; et al. A whole-genome association study of major determinants for host control of HIV-1. Science 2007, 317, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.G.; Marshall, E.E.; Malouli, D.; Ventura, A.B.; Hughes, C.M.; Ainslie, E.; Ford, J.C.; Morrow, D.; Gilbride, R.M.; Bae, J.Y.; et al. A live-attenuated RhCMV/SIV vaccine shows long-term efficacy against heterologous SIV challenge. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- McMichael, A.J.; Borrow, P.; Tomaras, G.D.; Goonetilleke, N.; Haynes, B.F. The immune response during acute HIV-1 infection: Clues for vaccine development. Nat. Rev. Immunol. 2010, 10, 11–23. [Google Scholar] [CrossRef]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Sun, J.C.; Bevan, M.J. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Wilson, N.S.; Waithman, J.; Villadangos, J.A.; Carbone, F.R.; Heath, W.R.; Belz, G.T. Cognate CD4+ T cell licensing of dendritic cells in CD8+ T cell immunity. Nat. Immunol. 2004, 5, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.R.; Carbone, F.R.; Karamalis, F.; Flavell, R.A.; Miller, J.F.; Heath, W.R. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature 1998, 393, 478–480. [Google Scholar] [CrossRef]
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.P.; Toes, R.E.; van der Voort, E.I.; Offringa, R.; Melief, C.J. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998, 393, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Bevan, M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, F.R.; Bevan, M.J. Class I-restricted processing and presentation of exogenous cell-associated antigen in vivo. J. Exp. Med. 1990, 171, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.Y.; Golumbek, P.; Ahmadzadeh, M.; Jaffee, E.; Pardoll, D.; Levitsky, H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science 1994, 264, 961–965. [Google Scholar] [CrossRef]
- Sigal, L.J.; Crotty, S.; Andino, R.; Rock, K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 1999, 398, 77–80. [Google Scholar] [CrossRef]
- Viola, A.; Lanzavecchia, A. T cell activation determined by T cell receptor number and tunable thresholds. Science 1996, 273, 104–106. [Google Scholar] [CrossRef]
- Tuosto, L.; Acuto, O. CD28 affects the earliest signaling events generated by TCR engagement. Eur. J. Immunol. 1998, 28, 2131–2142. [Google Scholar] [CrossRef]
- D’Souza, W.N.; Schluns, K.S.; Masopust, D.; Lefrancois, L. Essential role for IL-2 in the regulation of antiviral extralymphoid CD8 T cell responses. J. Immunol. 2002, 168, 5566–5572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.A.; Bevan, M.J. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007, 25, 171–192. [Google Scholar] [CrossRef] [PubMed]
- Sprent, J.; Surh, C.D. T cell memory. Annu. Rev. Immunol. 2002, 20, 551–579. [Google Scholar] [CrossRef]
- Meyerhoff, R.R.; Scearce, R.M.; Ogburn, D.F.; Lockwood, B.; Pickeral, J.; Kuraoka, M.; Anasti, K.; Eudailey, J.; Eaton, A.; Cooper, M.; et al. HIV-1 Consensus Envelope-Induced Broadly Binding Antibodies. Aids Res. Hum. Retrovir. 2017, 33, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Moyo, N.; Vogel, A.B.; Buus, S.; Erbar, S.; Wee, E.G.; Sahin, U.; Hanke, T. Efficient Induction of T Cells against Conserved HIV-1 Regions by Mosaic Vaccines Delivered as Self-Amplifying mRNA. Mol. Ther. Methods Clin. Dev. 2019, 12, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Ondondo, B.; Murakoshi, H.; Clutton, G.; Abdul-Jawad, S.; Wee, E.G.; Gatanaga, H.; Oka, S.; McMichael, A.J.; Takiguchi, M.; Korber, B.; et al. Novel Conserved-region T-cell Mosaic Vaccine With High Global HIV-1 Coverage Is Recognized by Protective Responses in Untreated Infection. Mol. Ther. 2016, 24, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Borthwick, N.; Ahmed, T.; Ondondo, B.; Hayes, P.; Rose, A.; Ebrahimsa, U.; Hayton, E.J.; Black, A.; Bridgeman, A.; Rosario, M.; et al. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol. Ther. 2014, 22, 464–475. [Google Scholar] [CrossRef] [Green Version]
- Moyo, N.; Wee, E.G.; Korber, B.; Bahl, K.; Falcone, S.; Himansu, S.; Wong, A.L.; Dey, A.K.; Feinberg, M.; Hanke, T. Tetravalent Immunogen Assembled from Conserved Regions of HIV-1 and Delivered as mRNA Demonstrates Potent Preclinical T-Cell Immunogenicity and Breadth. Vaccines (Basel) 2020, 8, 360. [Google Scholar] [CrossRef]
- Theiler, J.; Yoon, H.; Yusim, K.; Picker, L.J.; Fruh, K.; Korber, B. Epigraph: A Vaccine Design Tool Applied to an HIV Therapeutic Vaccine and a Pan-Filovirus Vaccine. Sci. Rep. 2016, 6, 33987. [Google Scholar] [CrossRef] [Green Version]
- Korber, B.; Fischer, W. T cell-based strategies for HIV-1 vaccines. Hum. Vaccin. Immunother. 2020, 16, 713–722. [Google Scholar] [CrossRef]
- Buchbinder, S.P.; Mehrotra, D.V.; Duerr, A.; Fitzgerald, D.W.; Mogg, R.; Li, D.; Gilbert, P.B.; Lama, J.R.; Marmor, M.; Del Rio, C.; et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): A double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 2008, 372, 1881–1893. [Google Scholar] [CrossRef] [Green Version]
- Hammer, S.M.; Sobieszczyk, M.E.; Janes, H.; Karuna, S.T.; Mulligan, M.J.; Grove, D.; Koblin, B.A.; Buchbinder, S.P.; Keefer, M.C.; Tomaras, G.D.; et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N. Engl. J. Med. 2013, 369, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ. Vaccines (Basel) 2013, 1, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- D’Haese, S.; Lacroix, C.; Garcia, F.; Plana, M.; Ruta, S.; Vanham, G.; Verrier, B.; Aerts, J.L. Off the beaten path: Novel mRNA-nanoformulations for therapeutic vaccination against HIV. J. Control. Release 2020. [Google Scholar] [CrossRef]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2020. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Alameh, M.G.; Weissman, D.; Pardi, N. Messenger RNA-Based Vaccines Against Infectious Diseases. Curr. Top. Microbiol. Immunol. 2020. [Google Scholar] [CrossRef]
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Kariko, K.; Buckstein, M.; Ni, H.P.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Kariko, K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 2019, 28. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Mir, F.F.; Jhunjhunwala, S.; Kaczmarek, J.C.; Hurtado, J.E.; Yang, J.H.; Webber, M.J.; Kowalski, P.S.; Heartlein, M.W.; DeRosa, F.; et al. Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials 2016, 109, 78–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Kariko, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic. Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic. Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, D.; Pardi, N.; Muramatsu, H.; Kariko, K. HPLC purification of in vitro transcribed long RNA. Methods Mol. Biol. 2013, 969, 43–54. [Google Scholar] [CrossRef]
- Zeng, Q.; Jiang, H.; Wang, T.; Zhang, Z.; Gong, T.; Sun, X. Cationic micelle delivery of Trp2 peptide for efficient lymphatic draining and enhanced cytotoxic T-lymphocyte responses. J. Control Release 2015, 200, 1–12. [Google Scholar] [CrossRef]
- Zhao, M.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2016, 23, 2596–2607. [Google Scholar] [CrossRef]
- Li, M.; Zhao, M.; Fu, Y.; Li, Y.; Gong, T.; Zhang, Z.; Sun, X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J. Control Release 2016, 228, 9–19. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175. [Google Scholar] [CrossRef]
- Coolen, A.L.; Lacroix, C.; Mercier-Gouy, P.; Delaune, E.; Monge, C.; Exposito, J.Y.; Verrier, B. Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 2019, 195, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007. [Google Scholar] [CrossRef]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef]
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombacz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Kariko, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther. Nucleic Acids 2019, 15, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Ni, H.; Scales, D.; Dude, A.; Capodici, J.; McGibney, K.; Abdool, A.; Isaacs, S.N.; Cannon, G.; Karikó, K. HIV gag mRNA transfection of dendritic cells (DC) delivers encoded antigen to MHC class I and II molecules, causes DC maturation, and induces a potent human in vitro primary immune response. J. Immunol. 2000, 165, 4710–4717. [Google Scholar] [CrossRef] [Green Version]
- Saeboe-Larssen, S.; Fossberg, E.; Gaudernack, G. mRNA-based electrotransfection of human dendritic cells and induction of cytotoxic T lymphocyte responses against the telomerase catalytic subunit (hTERT). J. Immunol. Methods 2002, 259, 191–203. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Boulassel, M.R.; Yassine-Diab, B.; Nicolette, C.; Healey, D.; Jain, R.; Landry, C.; Yegorov, O.; Tcherepanova, I.; Monesmith, T.; et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin. Immunol. 2010, 134, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Allard, S.D.; De Keersmaecker, B.; de Goede, A.L.; Verschuren, E.J.; Koetsveld, J.; Reedijk, M.L.; Wylock, C.; De Bel, A.V.; Vandeloo, J.; Pistoor, F.; et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin. Immunol. 2012, 142, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immun. Defic. Syndr. 2016, 71, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, J.M.; Routy, J.P.; Welles, S.; DeBenedette, M.; Tcherepanova, I.; Angel, J.B.; Asmuth, D.M.; Stein, D.K.; Baril, J.G.; McKellar, M.; et al. Dendritic Cell Immunotherapy for HIV-1 Infection Using Autologous HIV-1 RNA: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Jaids-J. Acq. Imm. Def. 2016, 72, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, C.L.; DeBenedette, M.A.; Tcherepanova, I.Y.; Gamble, A.; Lewis, W.E.; Cope, A.B.; Kuruc, J.D.; McGee, K.S.; Kearney, M.F.; Coffin, J.M.; et al. Immunogenicity of AGS-004 Dendritic Cell Therapy in Patients Treated During Acute HIV Infection. Aids Res. Hum. Retrovir. 2018, 34, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Guardo, A.C.; Joe, P.T.; Miralles, L.; Bargalló, M.E.; Mothe, B.; Krasniqi, A.; Heirman, C.; García, F.; Thielemans, K.; Brander, C.; et al. Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). Aids 2017, 31, 321–332. [Google Scholar] [CrossRef]
- Jong, W.; Leal, L.; Buyze, J.; Pannus, P.; Guardo, A.; Salgado, M.; Mothe, B.; Molto, J.; Moron-Lopez, S.; Gálvez, C.; et al. Therapeutic Vaccine in Chronically HIV-1-Infected Patients: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Trial with HTI-TriMix. Vaccines 2019, 7, 209. [Google Scholar] [CrossRef] [Green Version]
- Moyer, T.J.; Zmolek, A.C.; Irvine, D.J. Beyond antigens and adjuvants: Formulating future vaccines. J. Clin. Investig. 2016, 126, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Gause, K.T.; Wheatley, A.K.; Cui, J.; Yan, Y.; Kent, S.J.; Caruso, F. Immunological Principles Guiding the Rational Design of Particles for Vaccine Delivery. Acs Nano 2017, 11, 54–68. [Google Scholar] [CrossRef]
- Lopez-Sagaseta, J.; Malito, E.; Rappuoli, R.; Bottomley, M.J. Self-assembling protein nanoparticles in the design of vaccines. Comput. Struct. Biotechnol. J. 2016, 14, 58–68. [Google Scholar] [CrossRef] [Green Version]
- Sliepen, K.; Ozorowski, G.; Burger, J.A.; van Montfort, T.; Stunnenberg, M.; LaBranche, C.; Montefiori, D.C.; Moore, J.P.; Ward, A.B.; Sanders, R.W. Presenting native-like HIV-1 envelope trimers on ferritin nanoparticles improves their immunogenicity. Retrovirology 2015, 12, 82. [Google Scholar] [CrossRef] [Green Version]
- He, L.; de Val, N.; Morris, C.D.; Vora, N.; Thinnes, T.C.; Kong, L.; Azadnia, P.; Sok, D.; Zhou, B.; Burton, D.R.; et al. Presenting native-like trimeric HIV-1 antigens with self-assembling nanoparticles. Nat. Commun. 2016, 7, 12041. [Google Scholar] [CrossRef]
- Kariko, K.; Muramatsu, H.; Keller, J.M.; Weissman, D. Increased erythropoiesis in mice injected with submicrogram quantities of pseudouridine-containing mRNA encoding erythropoietin. Mol. Ther. 2012, 20, 948–953. [Google Scholar] [CrossRef] [Green Version]
- Bradley, T.; Kuraoka, M.; Yeh, C.H.; Tian, M.; Chen, H.; Cain, D.W.; Chen, X.; Cheng, C.; Ellebedy, A.H.; Parks, R.; et al. Immune checkpoint modulation enhances HIV-1 antibody induction. Nat. Commun. 2020, 11, 948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lederer, K.; Castaño, D.; Atria, D.G.; Oguin, T.H.; Wang, S.; Manzoni, T.B.; Muramatsu, H.; Hogan, M.J.; Amanat, F.; Cherubin, P.; et al. SARS-CoV-2 mRNA vaccines foster potent antigen-specific germinal center responses associated with neutralizing antibody generation. Immunity 2020. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, K.M.; Carnathan, D.G.; Nogal, B.; Martin, J.T.; Rodriguez, O.L.; Upadhyay, A.A.; Enemuo, C.A.; Gebru, E.H.; Choe, Y.; Viviano, F.; et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell 2020, 180, 206. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Pardi, N.; Parks, R.; Santra, S.; Mu, Z.; Sutherland, L.; Scearce, R.; Barr, M.; Eaton, A.; Hernandez, G.; et al. Lipid nanoparticle encapsulated nucleoside-modified mRNA vaccines elicit polyfunctional HIV-1 antibodies comparable to proteins in nonhuman primates. bioRxiv 2020. [Google Scholar] [CrossRef]
- Koup, R.A.; Douek, D.C. Vaccine design for CD8 T lymphocyte responses. Cold Spring Harb. Perspect Med. 2011, 1, a007252. [Google Scholar] [CrossRef] [Green Version]
- Barouch, D.H.; Picker, L.J. Novel vaccine vectors for HIV-1. Nat. Rev. Microbiol. 2014, 12, 765–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 induces SARS-CoV-2-neutralising antibodies and T cells in humans. medRxiv 2020. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gorman, J.; Geng, H.; Liu, Q.; Lin, Y.; Tsybovsky, Y.; Go, E.P.; Dey, B.; Andine, T.; Kwon, A.; et al. Interdomain Stabilization Impairs CD4 Binding and Improves Immunogenicity of the HIV-1 Envelope Trimer. Cell Host. Microbe. 2018, 23, 832–844.e836. [Google Scholar] [CrossRef] [Green Version]
- Henderson, R.; Lu, M.; Zhou, Y.; Mu, Z.; Parks, R.; Han, Q.; Hsu, A.L.; Carter, E.; Blanchard, S.C.; Edwards, R.J.; et al. Disruption of the HIV-1 Envelope allosteric network blocks CD4-induced rearrangements. Nat. Commun. 2020, 11, 520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.D.; Pancera, M.; Acharya, P.; Georgiev, I.S.; Crooks, E.T.; Gorman, J.; Joyce, M.G.; Guttman, M.; Ma, X.; Narpala, S.; et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat. Struct. Mol. Biol. 2015, 22, 522–531. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castells, M.C.; Phillips, E.J. Maintaining Safety with SARS-CoV-2 Vaccines. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbasi, J. COVID-19 and mRNA Vaccines-First Large Test for a New Approach. JAMA 2020, 324, 1125–1127. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu, Z.; Haynes, B.F.; Cain, D.W. HIV mRNA Vaccines—Progress and Future Paths. Vaccines 2021, 9, 134. https://doi.org/10.3390/vaccines9020134
Mu Z, Haynes BF, Cain DW. HIV mRNA Vaccines—Progress and Future Paths. Vaccines. 2021; 9(2):134. https://doi.org/10.3390/vaccines9020134
Chicago/Turabian StyleMu, Zekun, Barton F. Haynes, and Derek W. Cain. 2021. "HIV mRNA Vaccines—Progress and Future Paths" Vaccines 9, no. 2: 134. https://doi.org/10.3390/vaccines9020134
APA StyleMu, Z., Haynes, B. F., & Cain, D. W. (2021). HIV mRNA Vaccines—Progress and Future Paths. Vaccines, 9(2), 134. https://doi.org/10.3390/vaccines9020134