
Chimeric VLPs Based on HIV-1 Gag and a Fusion Rabies Glycoprotein Induce Specific Antibodies against Rabies and Foot-and-Mouth Disease Virus

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fusion Protein Construction. Plasmids
2.2. Cell Line, Media, and Culture Conditions
2.3. Transient Transfection
2.4. VLP Purification
2.5. Confocal Microscopy
2.5.1. Standard Confocal Laser Scanning Microscopy (CLSM)
2.5.2. Super-Resolution Fluorescence Microscopy (SRFM)
2.6. Flow Cytometry
2.7. VLPs Analysis and Quantitation
2.7.1. Bi-specific” Sandwich ELISA
2.7.2. Fluorescence-Based Quantification of VLP Production
2.7.3. Nanoparticle Tracking Analysis (NTA)
2.8. Electron Microscopy
2.8.1. Transmission Electron Microscopy (TEM)
2.8.2. Cryogenic Electron Microscopy (CryoTEM)
2.9. Mice Immunization and Serology
2.9.1. Immunization Plan
2.9.2. Quantification of RV Specific Immunoglobulins
2.9.3. Quantification of FMD Disease Epitope Specific Immunoglobulins
2.10. Statistical Analysis
3. Results
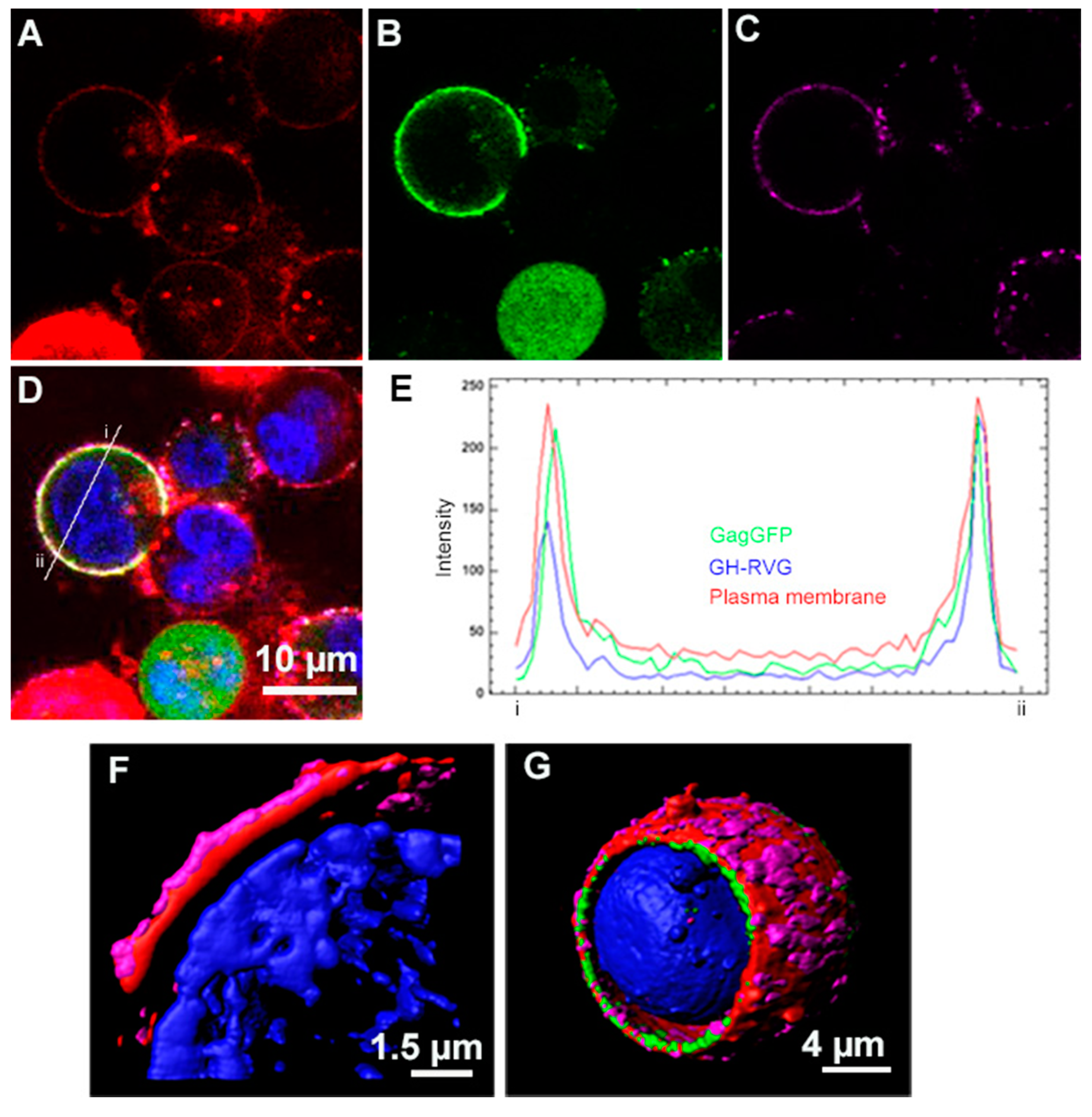
3.1. Design, Construction and Analysis of the Expression of GH-RVG Fusion Protein
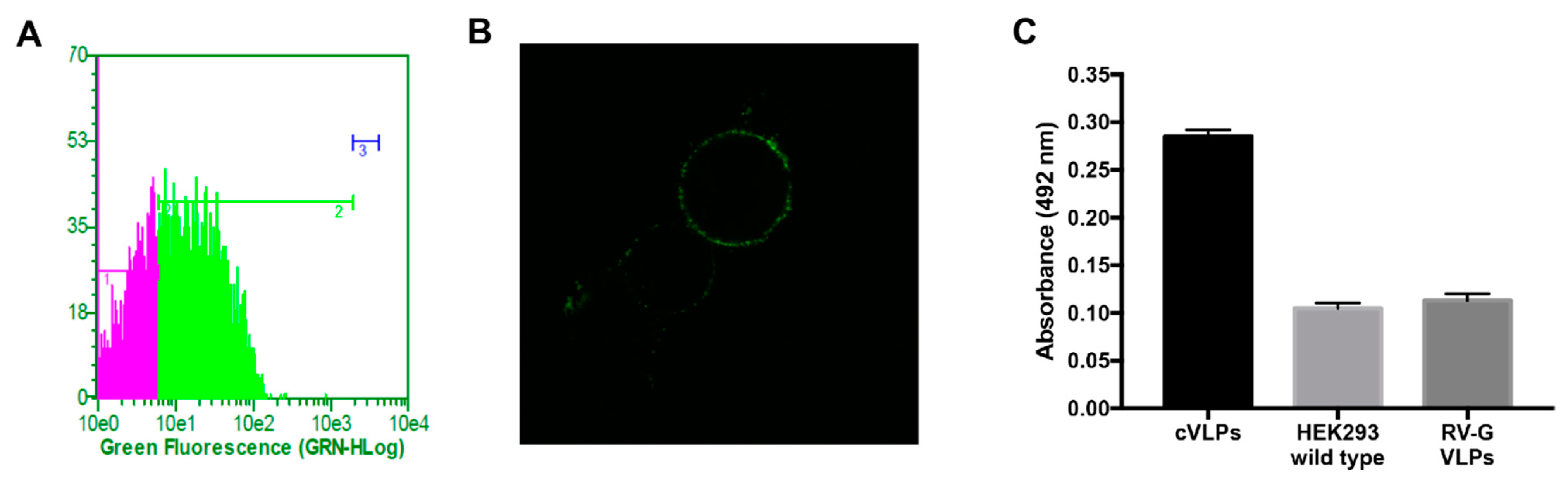
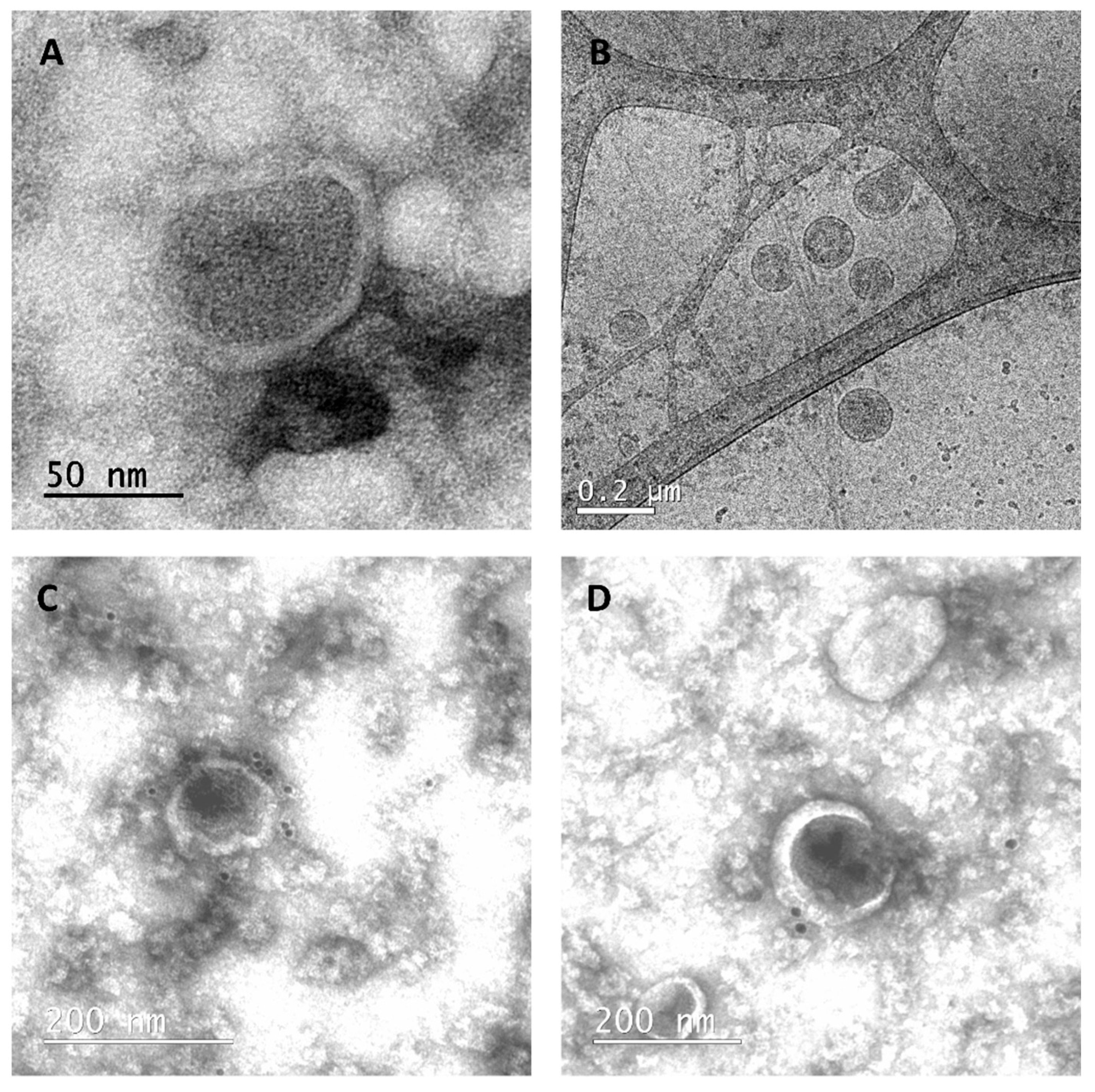
3.2. cVLP Expression and Characterization
3.3. cVLPs Production Optimization
3.4. Evaluation of cVLPs Immunogenicity in Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Azuma, H.; Yoneda, S. Structure and dynamics of the GH loop of the foot-and-mouth disease virus capsid. J. Mol. Graph. Model. 2009, 28, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Belsham, G.J. Towards improvements in foot-and-mouth disease vaccine performance. Acta Veter- Scand. 2020, 62, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bertona, D.; Pujato, N.; Bontempi, I.; Gonzalez, V.; Cabrera, G.; Gugliotta, L.; Hozbor, D.; Nicastro, A.; Calvinho, L.; Marcipar, I.S. Development and assessment of a new cage-like particle adjuvant. J. Pharm. Pharmacol. 2017, 69, 1293–1303. [Google Scholar] [CrossRef]
- Bidart, J.; Mignaqui, A.; Kornuta, C.; Lupi, G.; Gammella, M.; Soria, I.; Galarza, R.; Ferella, A.; Cardillo, S.; Langellotti, C.; et al. FMD empty capsids combined with the Immunostimulant Particle Adjuvant -ISPA or ISA206 induce protective immunity against Foot and Mouth Disease Virus. Virus Res. 2021, 198339, 198339. [Google Scholar] [CrossRef]
- Bidart, J.; Kornuta, C.; Gammella, M.; Gnazzo, V.; Soria, I.; Langellotti, C.; Mongini, C.; Galarza, R.; Calvinho, L.; Lupi, G.; et al. A New Cage-Like Particle Adjuvant Enhances Protection of Foot-and-Mouth Disease Vaccine. Front. Veter- Sci. 2020, 7, 396. [Google Scholar] [CrossRef]
- Bucafusco, D.; Di Giacomo, S.; Pega, J.; Schammas, J.M.; Cardoso, N.; Capozzo, A.V.; Perez-Filgueira, M. Foot-and-mouth disease vaccination induces cross-reactive IFN-γ responses in cattle that are dependent on the integrity of the 140S particles. Virology 2015, 476, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Burman, A.; Clark, S.; Abrescia, N.G.A.; Fry, E.E.; Stuart, D.I.; Jackson, T. Specificity of the VP1 GH Loop of Foot-and-Mouth Disease Virus for αv Integrins. J. Virol. 2006, 80, 9798–9810. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lu, Z.; Liu, Z. Foot-and-mouth disease vaccines: Progress and problems. Expert Rev. Vaccines 2016, 15, 783–789. [Google Scholar] [CrossRef]
- Cervera, L.; Fuenmayor, J.; González-Domínguez, I.; Gutiérrez-Granados, S.; Segura, M.M.; Gòdia, F. Selection and optimization of transfection enhancer additives for increased virus-like particle production in HEK293 suspension cell cultures. Appl. Microbiol. Biotechnol. 2015, 99, 9935–9949. [Google Scholar] [CrossRef]
- Cervera, L.; Gòdia, F.; Tarrés-freixas, F.; Aguilar-gurrieri, C.; Carrillo, J. Production of HIV-1-based virus-like particles for vacci-nation: Achievements and limits. Appl. Microbiol. Biotechnol. 2019, 103, 7367–7384. [Google Scholar] [CrossRef]
- Cervera, L.; González-Domínguez, I.; Segura, M.M.; Gòdia, F. Intracellular characterization of Gag VLP production by transient transfection of HEK 293 cells. Biotechnol. Bioeng. 2017, 114, 2507–2517. [Google Scholar] [CrossRef]
- Cervera, L.; Gutiérrez-Granados, S.; Martínez, M.; Blanco, J.; Gòdia, F.; Segura, M.M.; Martínez-Calle, M. Generation of HIV-1 Gag VLPs by transient transfection of HEK 293 suspension cell cultures using an optimized animal-derived component free medium. J. Biotechnol. 2013, 166, 152–165. [Google Scholar] [CrossRef] [PubMed]
- Chazal, N.; Gerlier, D. Virus Entry, Assembly, Budding, and Membrane Rafts. Microbiol. Mol. Biol. Rev. 2003, 67, 226–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christophe, P.; Stephanie, L.; Bernhard, D.; Monique, L. Glycoprotein of nonpathogenic rabies viruses is a key determinant of human cell apoptosis. J. Virol. 2003, 77, 10537–10547. [Google Scholar] [CrossRef]
- Crisci, E.; Bárcena, J.; Montoya, M. Virus-like particle-based vaccines for animal viral infections. Inmunología 2013, 32, 102–116. [Google Scholar] [CrossRef]
- Crisci, E.; Bárcena, J.; Montoya, M. Virus-like particles: The new frontier of vaccines for animal viral infections. Veter- Immunol. Immunopathol. 2012, 148, 211–225. [Google Scholar] [CrossRef]
- Segundo, F.D.-S.; Medina, G.N.; Stenfeldt, C.; Arzt, J.; Santos, T.D.L. Foot-and-mouth disease vaccines. Veter- Microbiol. 2017, 206, 102–112. [Google Scholar] [CrossRef]
- Doel, T.R.; Baccarini, P.J. Thermal stability of foot-and-mouth disease virus. Arch. Virol. 1981, 70, 21–32. [Google Scholar] [CrossRef]
- Donaldson, B.; Al-Barwani, F.; Young, V.; Scullion, S.; Ward, V.; Young, S. Virus-Like Particles, a Versatile Subunit Vaccine Platform. Adv. Deliv. Sci. Technol. 2014, 159–180. [Google Scholar] [CrossRef]
- Donaldson, B.; Lateef, Z.; Walker, G.F.; Young, S.L.; Ward, V.K. Virus-like particle vaccines: Immunology and formulation for clinical translation. Expert Rev. Vaccines 2018, 17, 833–849. [Google Scholar] [CrossRef]
- Fernandez-Sainz, I.; Gavitt, T.D.; Koster, M.; Ramirez-Medina, E.; Rodriguez, Y.Y.; Wu, P.; Silbart, L.K.; Santos, T.D.L.; Szczepanek, S.M. The VP1 G-H loop hypervariable epitope contributes to protective immunity against Foot and Mouth Disease Virus in swine. Vaccine 2019, 37, 3435–3442. [Google Scholar] [CrossRef]
- Fontana, D.; Kratje, R.; Etcheverrigaray, M.; Prieto, C. Immunogenic virus-like particles continuously expressed in mammalian cells as a veterinary rabies vaccine candidate. Vaccine 2015, 33, 4238–4246. [Google Scholar] [CrossRef]
- Fontana, D.; Kratje, R.; Etcheverrigaray, M.; Prieto, C. Rabies virus-like particles expressed in HEK293 cells. Vaccine 2014, 32, 2799–2804. [Google Scholar] [CrossRef]
- Fontana, D.; Marsili, F.; Etcheverrigaray, M.; Kratje, R.; Prieto, C. Rabies VLPs adjuvanted with saponin-based liposomes induce enhanced immunogenicity mediated by neutralizing antibodies in cattle, dogs and cats. J. Virol. Methods 2020, 286, 113966. [Google Scholar] [CrossRef]
- Fontana, D.; Rodriguez, M.C.; Garay, E.; Russo, S.; Prieto, C. Optimization and validation of a blocking ELISA for quantitation of anti-rabies immunoglobulins in multispecies sera. Appl. Microbiol. Biotechnol. 2020, 1–13. [Google Scholar] [CrossRef]
- Fuenmayor, J.; Cervera, L.; Gòdia, F.; Kamen, A. Extended gene expression for Gag VLP production achieved at bioreactor scale. J. Chem. Technol. Biotechnol. 2019, 94, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Fuenmayor, J.; Cervera, L.; Gutiérrez-Granados, S.; Gòdia, F. Transient gene expression optimization and expression vector comparison to improve HIV-1 VLP production in HEK293 cell lines. Appl. Microbiol. Biotechnol. 2017, 102, 165–174. [Google Scholar] [CrossRef]
- Fuenmayor, J.; Cervera, L.; Rigau, C.; Gòdia, F. Enhancement of HIV-1 VLP production using gene inhibition strategies. Appl. Microbiol. Biotechnol. 2018, 102, 4477–4487. [Google Scholar] [CrossRef]
- Fuenmayor, J.; Gòdia, F.; Cervera, L. Production of virus-like particles for vaccines. New Biotechnol. 2017, 39, 174–180. [Google Scholar] [CrossRef]
- González-Domínguez, I.; Gutiérrez-Granados, S.; Cervera, L.; Gòdia, F.; Domingo, N. Identification of HIV-1–Based Virus-like Particles by Multifrequency Atomic Force Microscopy. Biophys. J. 2016, 111, 1173–1179. [Google Scholar] [CrossRef] [Green Version]
- González-Domínguez, I.; Puente-Massaguer, E.; Cervera, L.; Gòdia, F. Quality Assessment of Virus-Like Particles at Single Particle Level: A Comparative Study. Viruses 2020, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- González-Domínguez, I.; Puente-Massaguer, E.; Cervera, L.; Gòdia, F. Quantification of the HIV-1 virus-like particle production process by super-resolution imaging: From VLP budding to nanoparticle analysis. Biotechnol. Bioeng. 2020, 117, 1929–1945. [Google Scholar] [CrossRef]
- Grubman, M.J.; Baxt, B. Foot-and-Mouth Disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-Granados, S.; Cervera, L.; Gòdia, F.; Carrillo, J.; Segura, M.M. Development and validation of a quantitation assay for fluorescently tagged HIV-1 virus-like particles. J. Virol. Methods 2013, 193, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Granados, S.; Cervera, L.; Kamen, A.A.; Gòdia, F. Advancements in mammalian cell transient gene expression (TGE) technology for accelerated production of biologics. Crit. Rev. Biotechnol. 2017, 38, 918–940. [Google Scholar] [CrossRef] [PubMed]
- Kurg, R.; Reinsalu, O.; Jagur, S.; Õunap, K.; Võsa, L.; Kasvandik, S.; Padari, K.; Gildemann, K.; Ustav, M. Biochemical and proteomic characterization of retrovirus Gag based microparticles carrying melanoma antigens. Sci. Rep. 2016, 6, 29425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavado-García, J.; Cervera, L.; Gòdia, F. An Alternative Perfusion Approach for the Intensification of Virus-Like Particle Production in HEK293 Cultures. Front. Bioeng. Biotechnol. 2020, 8, 617. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ge, S.; Li, L.; Wu, X.; Liu, Z.; Wang, Z. Virus-like particles: Potential veterinary vaccine immunogens. Res. Veter- Sci. 2012, 93, 553–559. [Google Scholar] [CrossRef]
- Liu, J.; Dai, S.; Wang, M.; Hu, Z.; Wang, H.; Deng, F. Virus like particle-based vaccines against emerging infectious disease viruses. Virol. Sin. 2016, 31, 279–287. [Google Scholar] [CrossRef] [Green Version]
- Lua, L.H.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P. Bioengineering virus-like particles as vaccines. Biotechnol. Bioeng. 2014, 111, 425–440. [Google Scholar] [CrossRef]
- Ludwig, C.; Wagner, R. Virus-like particles—universal molecular toolboxes. Curr. Opin. Biotechnol. 2007, 18, 537–545. [Google Scholar] [CrossRef]
- Mignaqui, A.C.; Ruiz, V.; Durocher, Y.; Wigdorovitz, A. Advances in novel vaccines for foot and mouth disease: Focus on recombinant empty capsids. Crit. Rev. Biotechnol. 2019, 39, 306–320. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Zha, L.; Cabral-Miranda, G.; Bachmann, M.F. Major findings and recent advances in virus–like particle (VLP)-based vaccines. Semin. Immunol. 2017, 34, 123–132. [Google Scholar] [CrossRef]
- Noad, R.; Roy, P. Virus-like particles as immunogens. Trends Microbiol. 2003, 11, 438–444. [Google Scholar] [CrossRef]
- Foot and Mouth Disease, by D. GEORGE COLLINS, Chairman, City of London Cattle Markets Committee. J. R. Sanit. Inst. 1913, 34, 132–136. [CrossRef]
- Rodriguez, L.L.; Grubman, M.J. Foot and mouth disease virus vaccines. Vaccine 2009, 27, D90–D94. [Google Scholar] [CrossRef]
- Roldão, A.; Silva, A.; Mellado, M.; Alves, P.; Carrondo, M. Viruses and Virus-Like Particles in Biotechnology: Fundamentals and Applications. In Reference Module in Life Sciences; Elsevier: Amsterdam, The Netherlands, 2017; pp. 633–656. [Google Scholar]
- Roldão, A.; Mellado, M.C.M.; Castilho, L.R.; Carrondo, M.J.T.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef]
- Santos, T.D.L.; Segundo, F.D.-S.; Rodriguez, L.L. The need for improved vaccines against foot-and-mouth disease. Curr. Opin. Virol. 2018, 29, 16–25. [Google Scholar] [CrossRef]
- Schnell, M.J.; Mcgettigan, J.P.; Wirblich, C.; Papaneri, A. The cell biology of rabies virus: Using stealth to reach the brain. Nat. Rev. Genet. 2009, 8, 51–61. [Google Scholar] [CrossRef]
- Singh, R.K.; Sharma, G.K.; Mahajan, S.; Dhama, K.; Basagoudanavar, S.H.; Hosamani, M.; Sreenivasa, B.P.; Chaicumpa, W.; Gupta, V.K.; Sanyal, A. Foot-and-Mouth Disease Virus: Immunobiology, Advances in Vaccines and Vaccination Strategies Addressing Vaccine Failures-An Indian Perspective. Vaccines 2019, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Soares, H.R.; Almeida, A.I.; Tomás, H.A.; Alves, P.M.; Coroadinha, A.S. Flexible pseudotyping of retrovirus using recombinase-mediated cassette exchange. Biotechnol. Lett. 2018, 40, 633–639. [Google Scholar] [CrossRef] [Green Version]
- Soares, H.R.; Castro, R.; Tomás, H.; Rodrigues, A.; Gomesalves, P.; Bellier, B.; Klatzmann, D.; Carrondo, M.J.T.; Coroadinha, A. Tetraspanins displayed in retrovirus-derived virus-like particles and their immunogenicity. Vaccine 2016, 34, 1634–1641. [Google Scholar] [CrossRef]
- Steppert, P.; Burgstaller, D.; Klausberger, M.; Tover, A.; Berger, E.; Jungbauer, A. Quantification and characterization of virus-like particles by size-exclusion chromatography and nanoparticle tracking analysis. J. Chromatogr. A 2017, 1487, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Venereo-Sanchez, A.; Simoneau, M.; Lanthier, S.; Chahal, P.; Bourget, L.; Ansorge, S.; Gilbert, R.; Henry, O.; Kamen, A. Process intensification for high yield production of influenza H1N1 Gag virus-like particles using an inducible HEK-293 stable cell line. Vaccine 2017, 35, 4220–4228. [Google Scholar] [CrossRef] [PubMed]
- Verdaguera, N.; Schoehnb, G.; Ochoa, W.F.; Fitaa, I.; Brookesc, S.; Kingc, A.; Domingod, E.; Mateu, M.G.; Stuart, D.I.; Hewat, E.A. Flexibility of the Major Antigenic Loop of Foot-and-Mouth Disease Virus Bound to a Fab Fragment of a Neutralising Antibody: Structure and Neutralisation. Virology 1999, 255, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO Expert Consultation on Rabies; World Health Organization Technical Report Series: Geneva, Switzerland, 2013. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fontana, D.; Garay, E.; Cervera, L.; Kratje, R.; Prieto, C.; Gòdia, F. Chimeric VLPs Based on HIV-1 Gag and a Fusion Rabies Glycoprotein Induce Specific Antibodies against Rabies and Foot-and-Mouth Disease Virus. Vaccines 2021, 9, 251. https://doi.org/10.3390/vaccines9030251
Fontana D, Garay E, Cervera L, Kratje R, Prieto C, Gòdia F. Chimeric VLPs Based on HIV-1 Gag and a Fusion Rabies Glycoprotein Induce Specific Antibodies against Rabies and Foot-and-Mouth Disease Virus. Vaccines. 2021; 9(3):251. https://doi.org/10.3390/vaccines9030251
Chicago/Turabian StyleFontana, Diego, Ernesto Garay, Laura Cervera, Ricardo Kratje, Claudio Prieto, and Francesc Gòdia. 2021. "Chimeric VLPs Based on HIV-1 Gag and a Fusion Rabies Glycoprotein Induce Specific Antibodies against Rabies and Foot-and-Mouth Disease Virus" Vaccines 9, no. 3: 251. https://doi.org/10.3390/vaccines9030251