Abstract
Cancer is the second leading cause of death worldwide. Today, the critical role of the immune system in tumor control is undisputed. Checkpoint antibody immunotherapy augments existing antitumor T cell activity with durable clinical responses in many tumor entities. Despite the presence of tumor-associated antigens and neoantigens, many patients have an insufficient repertoires of antitumor T cells. Autologous tumor vaccinations aim at alleviating this defect, but clinical success is modest. Loading tumor material into autologous dendritic cells followed by their laboratory expansion and therapeutic vaccination is promising, both conceptually and clinically. However, this process is laborious, time-consuming, costly, and hence less likely to solve the global cancer crisis. Therefore, it is proposed to re-focus on personalized anticancer vaccinations to enhance the immunogenicity of autologous therapeutic tumor vaccines. Recent work re-established the idea of using the alarming agents of the immune system, oxidative modifications, as an intrinsic adjuvant to broaden the antitumor T cell receptor repertoire in cancer patients. The key novelty is the use of gas plasma, a multi-reactive oxygen and nitrogen species-generating technology, for diversifying oxidative protein modifications in a, so far, unparalleled manner. This significant innovation has been successfully used in proof-of-concept studies and awaits broader recognition and implementation to explore its chances and limitations of providing affordable personalized anticancer vaccines in the future. Such multidisciplinary advance is timely, as the current COVID-19 crisis is inexorably reflecting the utmost importance of innovative and effective vaccinations in modern times.
1. Introduction
Each year 14.1 million new cases of cancers are diagnosed that require therapeutic attention. The classic pillars in oncology are surgery, radiotherapy, and chemotherapy. These measures have markedly improved median survival in patients across all types of cancer. However, significant progress has slowed down in the past decades for several reasons, radioresistance and chemoresistance being among them [1,2]. Meanwhile, biologicals, such as cytokines and antibodies targeting growth receptors, spurred therapy success [3,4,5]. A paradigm shift in oncology then came with the incorporation of antitumor immune defense into the treatment concepts and repertoires of the field of oncology. Although being predicted in the 1960s already [6,7], the concept needed several decades, and a leap in life science technology innovations, along with mechanistic concepts in immunology and oncology to harness its full potential. Today, antibodies targeting immunosuppressive checkpoint receptors on T cells have provided substantial clinical responses [8]. Their success, along with the Nobel Prize Award in Physiology and Medicine in 2018 for achievements in this field, has given antitumor T cells undisputed importance across the globe for providing tumor protection [9]. Tumor protection is carried out by generating antigen-specific T cells, followed by strengthening one’s immune response through a specific anti-tumor immune response. The personalized antigen vaccines have recently come to the fore [10] because they can be efficient and have few side effects. However, there are novel ways to optimize therapeutic anti-tumor vaccines in various strategies, such as a modified tumor biopsy vaccine [11], cryptic peptide [12], nano-particle loaded [13], or a PEG-modified antigen vaccine. Here we propose a new technical approach to optimize the immunization by mimicking a relevant biological process of the inflammatory microenvironment, namely the generation of reactive species.
2. Tumor Immune Evasion and Vaccination
Cancers evolve under the constant pressure of the immune system; a process called immune evasion [14]. Tumor variants with minimal activation of immune cells have a growth advantage over clonotypes with highly immunogenic antigens. This classic view was complemented over the last two decades with the opposite scenario. Highly immunogenic tumor cells do not attempt to hide from immune recognition but counteract immune cell activation by activating immunosuppressive ligands and receptors, for example, PD-L1, PD-L2, and CD80/86 [15]. Other mechanisms of an immunosuppressive microenvironment complement this camouflage and sabotage. For instance, hypoxia [16], soluble mediators such as kynurenine [17], and the promotion of suppressive immune cell subsets including M2 macrophages and regulator T cells [18]. However, the clinical success of checkpoint antibodies targeting receptors and ligands suggests the receptor-ligand-based immunosuppression of effector T cells as being a critical determinant of the therapeutic outcome. Hence, it is clear that strengthening the activity of existing antitumor T cell clones is a proven therapeutic concept in cancer immunotherapy.
A second complementary approach is broadening the T cell receptor repertoire by augmenting the generation of novel antitumor T cell clones. Autologous tumor vaccines provide a vast array of tumor-associated antigens (TAA) and neoantigens to the host. Such antigens are present in all types of tumors, albeit to a varying degree [19]. Another limitation is that not all of these tumor antigens are presentable on major histocompatibility complex (MHC) molecules due to the preference of protein digestion and peptide cleavage in the proteasome and immunoproteasome [20,21], as well as the affinity of the MHC receptor family towards specific amino acids of the peptide to provide suitable binding affinity [22,23]. However, the critical determinant of generating T cell activation or tolerance towards such antigens is the inflammatory context in which these are presented, along with the efficacy of antigen presentation. To address these issues, dendritic cells (DCs), being professional antigen-presenting cells, have been investigated in numerous preclinical and clinical studies for their ability to promote antitumor immunity after being loaded with tumor antigens in vitro [24,25]. Undoubtedly, this elegant type of cell therapy fostered the understanding of tumor immunology in oncology and benefited many patients enrolled in clinical trials. Nevertheless, this concept also has limitations. First, there was limited success in many clinical trials. Second, DC loading and expansion require state-of-the-art facilities and are associated with high costs. Even if near-ideal protocols had been, or were to be, developed, it is still questionable whether DC therapy would become a global gold standard for cancer therapy apart from in countries with privileged income and health care systems. Third, much focus has been put on DC activation and maturation. Simultaneously, the conditioning of the tumor material has received less attention, as was recently well demonstrated in a cohort of cancer patients [26], which at least gives rise to the idea of rethinking the inevitable need of DCs in the realm of tumor vaccination.
Textbook immunology predicts that the body has an inherent interest in mounting both B cell and T cell immune responses against (non-self) antigens if presented in a sufficiently inflammatory context. Adjuvants provide the latter, being the basis of vaccinations, a process currently receiving significant interest duringthe COVID-19 pandemic. Together, with the points mentioned above, this raises the question of what is limiting the use of autologous tumor material to be directly used as a vaccine without the need for external processing by other cell types. It is understood that early and recent attempts of using a native autologous tumor vaccination to provide therapeutic efficacy [27,28] failed. Notwithstanding, we here outline why reactive oxygen and nitrogen species might be a fascinating option to render tumor antigens more suitable for direct vaccination campaigns in oncology and possibly adjuvant to existing strategies [29,30,31,32], which are numerous and not covered here. It should be stressed that, in the tumor context, this text always refers to therapeutic vaccinations and not preventive/prophylactic vaccination.
3. Reactive Oxygen and Nitrogen Species
Reactive oxygen and nitrogen species (ROS/RNS) are molecules with great reactivity and abbreviated with ROS in this work, as most RNS contain oxygen. Besides their past underappreciation as mere metabolic byproducts, ROS are pivotal intracellular redox signaling agents [33], critical for infection control [34], and increasingly recognized as key elements of the inflammatory microenvironment. Immune cell activation and metabolic reprogramming of leukocyte subsets have been linked to endogenous ROS production as crucial to driving these processes [35,36,37,38]. Perhaps the best-known role of non-constitutive ROS is their early appearance during inflammation by immune cells and non-immune cells alike. ROS release is the very first event during tissue damage [39] and it is required for the subsequent neutrophil influx. Subsequent neutrophil priming and activation auto-amplifies ROS production, followed by another round of ROS amplification by incoming monocytes and macrophages that complement the reactive species array with several nitrogen species [40].
For instance, nitric oxide synthase (NOS) produces nitric oxide (NO), which reacts with superoxide (O2−), that is generated by NADPH (nicotinamide adenine dinucleotide phosphate) oxidases (NOX), to yield peroxynitrite (-ONOO). The enzyme superoxide dismutase (SOD) catalyzes the reaction of superoxide to hydrogen peroxide (H2O2). In the presence of hydrogen peroxide, the arterial indoleamine 2,3-dioxygenase 1 (IDO-1) formates singlet oxygen (1O2) for blood pressure regulation and vascular tone during inflammation [41]. In the presence of iron, H2O2 promotes the generation of highly reactive hydroxyl radicals (HO.) in the Fenton reaction [42]. Furthermore, myeloperoxidase (MPO) is known to generate hypobromous acid, hypochlorous acid, and hypothiocyanite. The hypochlorite radicals can participate in the formation of atomic oxygen (O) and HO [34]. This is the environment in which infection-related antigens are recognized, modified, and transported to the secondary lymphatic organs to activate adaptive immunity.
Current vaccine preparation strategies almost unanimously neglect this ancient evolutionary part of antigen modification. When taking a view into other research fields, this comes as a surprise. For decades, researchers have identified a pivotal role of ROS and oxidative post-translational modifications (oxPTMs) in autoimmunity [43]. Chronic inflammation and chronic ROS release modified antigens, leading to auto-antibodies and auto-reactive T cells that are observed in numerous diseases, including rheumatoid arthritis, systemic lupus erythematosus, and diabetes [44,45,46,47,48], partly in a neoepitope-like fashion [49,50]. Mechanistically, oxPTMs have been ascribed a function similar to damage-associated molecular patterns (DAMPs) [51], providing pro-inflammatory stimuli in in professional antigen-presenting cells (APCs), and are decisive for the balance between antigen tolerance and immunity. Altogether, multiple ROS modify antigens, leading to a DAMP-like character to activate innate immunity and potentially neoepitopes to broaden adaptive immunity and the B cell and T cell receptor repertoire. ROS are, therefore, ideal candidates to increase the immunogenicity of autologous tumor vaccines. However, the challenges of working with ROS are numerous. First, their production, reaction kinetics, and specificity are hard to control, apart from the short half-lives associated with most species. Second, oxidative modifications are challenging to track and require sophisticated infrastructure and bioinformatics for their analysis. Third, and most notably, a simultaneous generation of several highly reactive compounds is technically impossible unless utilizing a concept from physics: gas plasma technology.
4. Gas Plasma Technology as a Significant Innovation in Generating Multi-ROS Cocktails
Gas plasma is an electron-impact and photon-driven technology. In gas plasma jets, usually, a noble gas is excited by a high-frequency electrode [52]. Excited noble gas species transfer their chemical energy to oxygen and nitrogen in the ambient air, generating vast amounts of several reactive oxygen and nitrogen species simultaneously. Compared to hot gas plasma, cold plasmas are operated at body temperatures and therefore do not denature proteins or harm cells and tissue by thermal energy transfer. Therefore, the main product is the bio-active multi-ROS cocktail [53,54,55]. Similar to the ROS released during inflammation, plasmas generate short-lived species (O, •NO, •NO, O2•-, •OOH, -ONOO, 1O2, etc.) as well as long-lived molecules that are mostly deterioration products from short-lived species such as H2O2, NO2−, NO3−, and HOCl [56,57]. Hundreds of chemical reactions have been identified in gas plasma jets using computer modeling, and redox biology currently does not offer the tools to identify each of the reaction products unambiguously. The degree of complexity is increased when considering the different spatio-temporal concentrations of each of the species along the axis of a plasma jet. Nevertheless, gas plasmas are unique in their ability to deliver multi-ROS cocktails onto biologically relevant targets. Strikingly, the ROS cocktail can be modified by changing the gas composition fed into the plasma jets (Figure 1). This leads to an enrichment of some types of ROS and a partial depletion of others [58,59]. This way, unique oxidative modification patterns are being generated at biological target molecules, as recently shown for the model peptide cysteine using mass spectrometry [60]. Additionally, prototypic plasma jets often allow other parameters to be tuned, for instance, the feed gas flux, the excitation frequency and wave form, and the input power. Other studies confirmed the modification of antigens and proteins by plasmas [61,62], leading to functional changes [63,64,65].
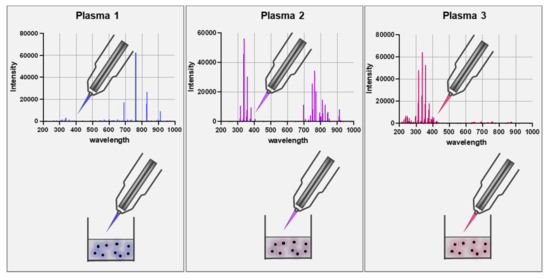
Figure 1.
Scheme of using different feed gas settings to generate gas plasma with distinct ROS cocktail profiles. The upper panel represents optical emission spectroscopy (intensity: relative units; wavelength: nanometer) measurements of the visible plasma effluent leaving the jet device. The lower panel is a schematic of a biological target being exposed to the gas plasma resulting in distinct oxidative modification patterns as depicted with the color code.
5. Proof-of-Concept Study Using Multi-ROS Cocktails to Provide Vaccine Tumor Control
In a recent study, we used chicken ovalbumin (Ova) to study the immunogenicity of multi-ROS cocktails in vitro and in vivo [66]. Using transgenic OT-II mice harboring Ova-specific T cells, we found gas plasma modified Ova to elicit significantly enhanced T cell activation compared to native antigen (Ova). This effect was specific, as it could not be replicated using human albumin. Splenocytes of other mice strains also did not show any elevated T cell activation. Strikingly, the enhanced T cell activation seen with gas plasma-treated Ova was not recapitulated when modifying Ova with equimolar amounts of long-lived reaction products from the plasma in treated liquids (H2O2, NO2−, NO3−, HOCl), unambiguously pointing towards a role of the unique cocktails generated by short-lived species. One plasma condition had more substantial effects than another one, which—in this specific setting—suggested a role of singlet and atomic oxygen or, possibly, lower ozone levels in providing immunogenic oxidative modifications. Using mass spectrometry, dozen of different modifications (e.g., oxidation, dioxidation, trioxidation, chlorination, and quinones) were found at many of the over 400 amino acids. This exemplifies the high degree of complexity, especially when considering the multi-ROS nature of the gas plasma system, currently making it difficult to come to a specific conclusion on which modifications have what effect. Some modifications were also in the sequence of the cognate peptide region. Moreover, it is possible (and likely, in our hands) that our observations were not based on one single type of modification or amino acid, but were rather a result of several modifications, complicating the control and understanding of this tool, as of now. We also found that oxidatively modified full Ova protein was needed, as the treatment of the oxidated immunogenic peptide alone (27 amino acids) did not elevate T cell responses. All this notwithstanding, the in vivo findings clearly showed functional consequences of the multi-ROS exposure to the antigen. In naïve mice, an increased anti-Ova T cell activity was created when using oxidized over native Ova, which was also reflected in a more inflammatory cytokine release profile. Notably, the gas plasma-derived multi-ROS Ova antigen oxidative modification led to significantly decreased tumor growth of Ova-expressing melanoma cells when given as a vaccine in a prime-boost scheme, compared to native untreated Ova. This was accompanied by the higher numbers and activation profiles of intratumoral T cells. These results emphasize the power of the multi-ROS antigen modification concept.
6. Concept and Challenges of Multi-ROS-Modified Autologous Tumor Vaccines
We propose gas plasma technology to upgrade antitumor vaccines by increasing adjuvanticity and antigenicity: the former due to the DAMP character of antigen oxPTMs promoting DC activation, as in the concept of immunogenic cell death (ICD) [67,68]. Indeed, gas plasma technology was shown to induce ICD [69], change proteomics suggesting neoepitope presentation [70], and increase the activity of antigen-presenting cells [71]. The increased immunogenicity of oxPTM and the potential formation of neoantigens was observed in autoimmunity [47,48]. However, the development of autoimmune disorders is not always based on oxidized antigens. Instead, some antigens are native [72], citrullinated [73,74] or deaminated [75]. Interestingly, Hultqvist and colleagues have shown that elevating the low oxidative burst capacity led to suppressing an autoimmune response [76,77]. With gas plasma technology, we mimic the ROS production of an oxidative burst to modify tumor-associated antigens.
We propose to homogenize collected autologous tumor tissue, followed by gas plasma exposure of the tumor lysates in a defined and pre-optimized setting (Figure 2).
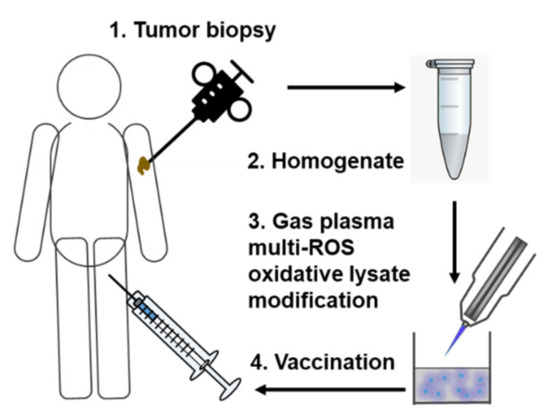
Figure 2.
Simplified scheme of gas plasma technology-mediated multi-ROS-driven improvement of autologous tumor vaccines. After tumor biopsy and homogenization of the tumor material, oxidation with complex multi-ROS cocktails generated by medical gas plasma jet technology follows prior to vaccination.
The lysates may be stored frozen in several aliquots until used in multiple vaccination rounds. Next, the multi-ROS oxidized homogenates can be thawed and combined with a pre-optimized adjuvant; a process that could be performed in a quality-controlled environment, such as pharmacies. In a series of elegant studies, the team of Lana Kandalaft employed HOCl-oxidized whole tumor lysates fed to autologous DCs, followed by the therapeutic vaccination of the latter, in a cohort of ovarian cancer patients [26]. Hundreds of vaccines generated in this way were well-tolerated without serious side effects. This study, which has been preceded by a decade of research [78,79,80,81,82,83,84,85,86], is the clinical proof-of-concept that oxidation of autologous tumor antigen potentiates antitumor immunity. Since, in our setting, optimized gas plasma exposure was even enhanced compared to the effect of HOCl, an additional benefit of multi-ROS modification might be feasible. So far, there have been three studies using gas plasma-inactivated tumor cells as a preventive/prophylactic vaccine that significantly decreased the tumor growth of live tumor cells given 7–9 days after vaccination [87,88,89]. Elevated tumor-infiltrated T cells with memory phenotypes were shown in plasma-treated tumors, and in patients, the infiltrated immune cells correlated with a better outcome [90,91]. As mechanisms of action, ICD was also recently proposed to improve DC antitumor vaccinations [92,93]. Further motivating our approach, there have been promising results with oxidized mannan-MUC1 (Mucin 1, cell surface associated; CD227) vaccination, as concluded from a 15-year follow-up study showing significantly fewer recurrences [94].
It is understood that there are many degrees of freedom in this concept. The ideal ROS cocktail needs to be identified. The gas plasma technology exposure needs to be implemented in a quality-managed environment. Either the necessity of DCs or the ideal adjuvant needs to be established, along with questions on optimized absolute dosing, injection frequency, administration routes, and potential deterioration of the ROS-treated vaccine. There might also be interdependence between these parameters. For instance, it was recently reported that a sequential intravenous priming vaccination followed by a later intradermal boost vaccination showed the best effects in providing antitumor immunity in mice, while simultaneous administration significantly worsened the outcome [95]. Apart from these practical aspects, the scientific challenges lie in the mapping of the type and number of the oxidative antigen modifications, the identification of optimal ROS cocktails for maximizing immunogenicity, the elucidation of putative APC receptors needed to recognize oxidized antigen, and the clarification of the role of the proteasomal and antigen-presenting machinery activity to stimulate cognate antigen recognition optimally in the host. The T cell activation and differentiation are dependent on the binding affinity between epitope and MHC-molecules and between MHC-complex and T cell receptors [96,97]. Consequently, the presentation of oxidatively modified antigens can alter binding affinities [74], possibly resulting in an increase or even decrease in T cell activation. After all, even a presumably perfect antitumor vaccine cannot circumvent tumor microenvironments that are hostile or suppressive to T cells. Therefore, vaccination should be embedded in a treatment strategy that also addresses these challenges. Finally, augmenting antitumor immunity, especially when using whole tumor lysates containing mostly self-antigens, is always at the verge of promoting autoimmunity. However, at least in the trials performed by Kandalaft and colleagues, such adverse events were not observed [81].
7. Conclusions
Therapeutic autologous antitumor vaccination is an elegant way of providing personalized therapy in oncology. However, its efficacy and practicability are limited by different constraints, and new enhanced vaccine technologies are required. Due to the current lack of validated biomarkers and neoantigens, upgrading a biopsy of the tumor is an intelligent, time-saving, and cost-effective way to enable personalized therapy. Oxidizing and modifying autologous tumor material with multiple reactive oxygen and nitrogen species simultaneously seems a promising avenue to increase both antigenicity (enhanced T cell receptor repertoire) and immunogenicity (increased co-stimulation and activation of adaptive antitumor immunity). Medical gas plasma jet technology is a recent innovation capable of providing multi-ROS cocktails in a unique and equivocal manner. Here, it is proposed to consider implementing this novel tool and to advocate its potential and limitations in providing efficient, fast, and affordable autologous antitumor vaccination, not just to those in privileged health care systems.
Author Contributions
Conceptualization, S.B. and R.C.; methodology, S.B. and R.C.; resources, S.B.; writing—original draft preparation, S.B. and R.C.; writing—review and editing, S.B. and R.C.; visualization, S.B. and R.C.; supervision, S.B.; project administration, S.B.; funding acquisition, S.B. All authors have read and agreed to the published version of the manuscript.
Funding
The work within the research group ZIK plasmatis “Plasma-Redox-Effects” is funded by the German Federal Ministry of Education and Research (BMBF, grant numbers 03Z22DN11, 03Z22Di1, 03Z22D511, 03COV06A, and 16GW0344K), the European Social Fund (ESF) conjoint with the Ministry of Education, Science, and Culture of Mecklenburg-Vorpommern, Germany (grant number ESF/14-BM-A55-0006-18), the German Research Foundation (DFG, grant number AOBJ 669606), the Stiftung Tumorforschung Kopf-Hals, the Ferdinand-Eisenberger Stiftung (grant number GeN1/FE-20), the Gerhard-Domagk Foundation Greifswald, and the Comprehensive Cancer Center Mecklenburg-Vorpommern (CCC-MV) seed funding initiative at the Greifswald University Medical Center in Greifswald, Germany. The funding sources had no role in the design of this study or its execution, analyses, interpretation of the data, or decision to publish the results.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Ghetie, M.-A.; Ghetie, V.; Vitetta, E.S. Section Review Biologicals & Immunologicals: The use of immunoconjugates in cancer therapy. Expert Opin. Investig. Drugs 2008, 5, 309–321. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Piessens, W.F. Evidence for human cancer immunity. A review. Cancer 1970, 26, 1212–1220. [Google Scholar] [CrossRef]
- Finney, J.W.; Byers, E.H.; Wilson, R.H. Studies in Tumor Auto-Immunity. Cancer Res. 1960, 20, 351–356. [Google Scholar]
- Azoury, S.C.; Straughan, D.M.; Shukla, V. Immune Checkpoint Inhibitors for Cancer Therapy: Clinical Efficacy and Safety. Curr. Cancer Drug Targets 2015, 15, 452–462. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Wang, T.; Wang, D.; Yu, H.; Feng, B.; Zhou, F.; Zhang, H.; Zhou, L.; Jiao, S.; Li, Y. A cancer vaccine-mediated postoperative immunotherapy for recurrent and metastatic tumors. Nat. Commun. 2018, 9, 1532. [Google Scholar] [CrossRef]
- Kotsakis, A.; Vetsika, E.K.; Christou, S.; Hatzidaki, D.; Vardakis, N.; Aggouraki, D.; Konsolakis, G.; Georgoulias, V.; Christophyllakis, C.; Cordopatis, P.; et al. Clinical outcome of patients with various advanced cancer types vaccinated with an optimized cryptic human telomerase reverse transcriptase (TERT) peptide: Results of an expanded phase II study. Ann. Oncol. 2012, 23, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Goldinger, S.M.; Dummer, R.; Baumgaertner, P.; Mihic-Probst, D.; Schwarz, K.; Hammann-Haenni, A.; Willers, J.; Geldhof, C.; Prior, J.O.; Kundig, T.M.; et al. Nano-particle vaccination combined with TLR-7 and -9 ligands triggers memory and effector CD8(+) T-cell responses in melanoma patients. Eur. J. Immunol. 2012, 42, 3049–3061. [Google Scholar] [CrossRef]
- Topfer, K.; Kempe, S.; Muller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef]
- Poschke, I.; Mougiakakos, D.; Kiessling, R. Camouflage and sabotage: Tumor escape from the immune system. Cancer Immunol. Immunother. 2011, 60, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 138, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Dalet, A.; Stroobant, V.; Vigneron, N.; Van den Eynde, B.J. Differences in the production of spliced antigenic peptides by the standard proteasome and the immunoproteasome. Eur. J. Immunol. 2011, 41, 39–46. [Google Scholar] [CrossRef]
- Van den Eynde, B.T.J.; Morel, S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr. Opin. Immunol. 2001, 13, 147–153. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Kloetzel, P.M. The proteasome and MHC class I antigen processing. Biochim. Biophys. Acta 2004, 1695, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex vivo pulsed dendritic cell vaccination against cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Farkas, A.; Conrad, C. Dendritic-cell-based therapeutic vaccination against cancer. Curr. Opin. Immunol. 2005, 17, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R.; Baumgartner, P.; Stevenson, B.J.; Iseli, C.; Dangaj, D.; et al. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Olson, K.B.; Laffin, R.; Horton, J.; Sullivan, J. Treatment of advanced cancer with active immunization. Cancer 1969, 24, 932–937. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Jahan, T.; Ross, H.; Sterman, D.; Richards, D.; Fox, B.; Jablons, D.; Aimi, J.; Lin, A.; Hege, K. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006, 13, 555–562. [Google Scholar] [CrossRef]
- Saini, R.; Lee, N.V.; Liu, K.Y.; Poh, C.F. Prospects in the Application of Photodynamic Therapy in Oral Cancer and Premalignant Lesions. Cancers 2016, 8, 83. [Google Scholar] [CrossRef]
- Schwaab, T.; Tretter, C.; Gibson, J.J.; Cole, B.F.; Schned, A.R.; Harris, R.; Wallen, E.M.; Fisher, J.L.; Waugh, M.G.; Truman, D.; et al. Immunological effects of granulocyte-macrophage colony-stimulating factor and autologous tumor vaccine in patients with renal cell carcinoma. J. Urol. 2004, 171, 1036–1042. [Google Scholar] [CrossRef]
- Olin, M.R.; Pluhar, G.E.; Andersen, B.M.; Shaver, R.; Waldron, N.N.; Moertel, C.L. Victory and defeat in the induction of a therapeutic response through vaccine therapy for human and canine brain tumors: A review of the state of the art. Crit. Rev. Immunol. 2014, 34, 399–432. [Google Scholar] [CrossRef]
- Li, L.; Ma, B.; Wang, W. Peptide-Based Nanomaterials for Tumor Immunotherapy. Molecules 2020, 26, 132. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins—Molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brustle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef]
- Rashida Gnanaprakasam, J.N.; Wu, R.; Wang, R. Metabolic Reprogramming in Modulating T Cell Reactive Oxygen Species Generation and Antioxidant Capacity. Front. Immunol. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef]
- Stanley, C.P.; Maghzal, G.J.; Ayer, A.; Talib, J.; Giltrap, A.M.; Shengule, S.; Wolhuter, K.; Wang, Y.; Chadha, P.; Suarna, C.; et al. Singlet molecular oxygen regulates vascular tone and blood pressure in inflammation. Nature 2019, 566, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Hirshberg, J.; Lyle, D.; Freij, J.B.; Caturegli, P. Reactive oxygen species in organ-specific autoimmunity. Auto Immun Highlights 2016, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, H.R. Is the generation of neo-antigenic determinants by free radicals central to the development of autoimmune rheumatoid disease? Autoimmun. Rev. 2008, 7, 544–549. [Google Scholar] [CrossRef]
- Arif, Z.; Neelofar, K.; Tarannum, A.; Arfat, M.Y.; Ahmad, S.; Zaman, A.; Khan, M.A.; Badar, A.; Islam, S.N.; Iqubal, M.A. SLE autoantibodies are well recognized by peroxynitrite-modified-HSA: Its implications in the pathogenesis of SLE. Int. J. Biol. Macromol. 2018, 106, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Autoimmunity and oxidatively modified autoantigens. Autoimmun. Rev. 2008, 7, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Nybo, T.; Dieterich, S.; Gamon, L.F.; Chuang, C.Y.; Hammer, A.; Hoefler, G.; Malle, E.; Rogowska-Wrzesinska, A.; Davies, M.J. Chlorination and oxidation of the extracellular matrix protein laminin and basement membrane extracts by hypochlorous acid and myeloperoxidase. Redox Biol. 2019, 20, 496–513. [Google Scholar] [CrossRef]
- Strollo, R.; Vinci, C.; Arshad, M.H.; Perrett, D.; Tiberti, C.; Chiarelli, F.; Napoli, N.; Pozzilli, P.; Nissim, A. Antibodies to post-translationally modified insulin in type 1 diabetes. Diabetologia 2015, 58, 2851–2860. [Google Scholar] [CrossRef]
- Mannering, S.I.; Di Carluccio, A.R.; Elso, C.M. Neoepitopes: A new take on beta cell autoimmunity in type 1 diabetes. Diabetologia 2019, 62, 351–356. [Google Scholar] [CrossRef]
- Yang, M.L.; Doyle, H.A.; Clarke, S.G.; Herold, K.C.; Mamula, M.J. Oxidative Modifications in Tissue Pathology and Autoimmune Disease. Antioxid. Redox Signal. 2018, 29, 1415–1431. [Google Scholar] [CrossRef]
- Carta, S.; Castellani, P.; Delfino, L.; Tassi, S.; Vene, R.; Rubartelli, A. DAMPs and inflammatory processes: The role of redox in the different outcomes. J. Leukoc. Biol. 2009, 86, 549–555. [Google Scholar] [CrossRef]
- Winter, J.; Brandenburg, R.; Weltmann, K.D. Atmospheric pressure plasma jets: An overview of devices and new directions. Plasma Sources Sci. Technol. 2015, 24, 064001. [Google Scholar] [CrossRef]
- Privat-Maldonado, A.; Schmidt, A.; Lin, A.; Weltmann, K.D.; Wende, K.; Bogaerts, A.; Bekeschus, S. ROS from Physical Plasmas: Redox Chemistry for Biomedical Therapy. Oxid. Med. Cell. Longev. 2019, 2019, 9062098. [Google Scholar] [CrossRef]
- Graves, D.B. Mechanisms of Plasma Medicine: Coupling Plasma Physics, Biochemistry, and Biology. IEEE Trans. Radiat. Plasma Med. Sci. 2017, 1, 281–292. [Google Scholar] [CrossRef]
- Von Woedtke, T.; Schmidt, A.; Bekeschus, S.; Wende, K.; Weltmann, K.D. Plasma Medicine: A Field of Applied Redox Biology. In Vivo 2019, 33, 1011–1026. [Google Scholar] [CrossRef]
- Wende, K.; von Woedtke, T.; Weltmann, K.D.; Bekeschus, S. Chemistry and biochemistry of cold physical plasma derived reactive species in liquids. Biol. Chem. 2018, 400, 19–38. [Google Scholar] [CrossRef]
- Schmidt-Bleker, A.; Winter, J.; Iseni, S.; Dunnbier, M.; Weltmann, K.D.; Reuter, S. Reactive species output of a plasma jet with a shielding gas device-combination of FTIR absorption spectroscopy and gas phase modelling. J. Phys. D Appl. Phys. 2014, 47, 145201. [Google Scholar] [CrossRef]
- Jablonowski, H.; Santos Sousa, J.; Weltmann, K.D.; Wende, K.; Reuter, S. Quantification of the ozone and singlet delta oxygen produced in gas and liquid phases by a non-thermal atmospheric plasma with relevance for medical treatment. Sci. Rep. 2018, 8, 12195. [Google Scholar] [CrossRef]
- Jablonowski, H.; Schmidt-Bleker, A.; Weltmann, K.D.; von Woedtke, T.; Wende, K. Non-touching plasma-liquid interaction—Where is aqueous nitric oxide generated? Phys. Chem. Chem. Phys. 2018, 20, 25387–25398. [Google Scholar] [CrossRef]
- Lackmann, J.W.; Wende, K.; Verlackt, C.; Golda, J.; Volzke, J.; Kogelheide, F.; Held, J.; Bekeschus, S.; Bogaerts, A.; Schulz-von der Gathen, V.; et al. Chemical fingerprints of cold physical plasmas—An experimental and computational study using cysteine as tracer compound. Sci. Rep. 2018, 8, 7736. [Google Scholar] [CrossRef]
- Wenske, S.; Lackmann, J.-W.; Bekeschus, S.; Weltmann, K.-D.; von Woedtke, T.; Wende, K. Nonenzymatic post-translational modifications in peptides by cold plasma-derived reactive oxygen and nitrogen species. Biointerphases 2020, 15. [Google Scholar] [CrossRef] [PubMed]
- Yusupov, M.; Lackmann, J.-W.; Razzokov, J.; Kumar, S.; Stapelmann, K.; Bogaerts, A. Impact of plasma oxidation on structural features of human epidermal growth factor. Plasma Process. Polym. 2018, 15. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, J.; Shen, J.; Lan, Y.; Ding, L.; Qian, S.; Cheng, C.; Xia, W.; Chu, P.K. Comparison of the Effects Induced by Plasma Generated Reactive Species and H2O2 on Lactate Dehydrogenase (LDH) Enzyme. IEEE Trans. Plasma Sci. 2018, 46, 2742–2752. [Google Scholar] [CrossRef]
- Krewing, M.; Stepanek, J.J.; Cremers, C.; Lackmann, J.W.; Schubert, B.; Muller, A.; Awakowicz, P.; Leichert, L.I.O.; Jakob, U.; Bandow, J.E. The molecular chaperone Hsp33 is activated by atmospheric-pressure plasma protecting proteins from aggregation. J. R. Soc. Interface 2019, 16, 20180966. [Google Scholar] [CrossRef]
- Krewing, M.; Jung, C.T.K.; Dobbelstein, E.; Schubert, B.; Jacob, T.; Bandow, J.E. Dielectric barrier discharge plasma treatment affects stability, metal ion coordination, and enzyme activity of bacterial superoxide dismutases. Plasma Process. Polym. 2020, 17. [Google Scholar] [CrossRef]
- Clemen, R.; Freund, E.; Mrochen, D.; Miebach, L.; Schmidt, A.; Rauch, B.H.; Lackmann, J.W.; Martens, U.; Wende, K.; Lalk, M.; et al. Gas Plasma Technology Augments Ovalbumin Immunogenicity and OT-II T Cell Activation Conferring Tumor Protection in Mice. Adv. Sci. 2021. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Khalili, M.; Daniels, L.; Lin, A.; Krebs, F.C.; Snook, A.E.; Bekeschus, S.; Bowne, W.B.; Miller, V. Non-Thermal Plasma-Induced Immunogenic Cell Death in Cancer: A Topical Review. J. Phys. D Appl. Phys. 2019, 52. [Google Scholar] [CrossRef]
- De Backer, J.; Razzokov, J.; Hammerschmid, D.; Mensch, C.; Hafideddine, Z.; Kumar, N.; van Raemdonck, G.; Yusupov, M.; Van Doorslaer, S.; Johannessen, C.; et al. The effect of reactive oxygen and nitrogen species on the structure of cytoglobin: A potential tumor suppressor. Redox Biol. 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Bekeschus, S.; Ressel, V.; Freund, E.; Gelbrich, N.; Mustea, A.; Stope, M.B. Gas Plasma-Treated Prostate Cancer Cells Augment Myeloid Cell Activity and Cytotoxicity. Antioxidants 2020, 9, 323. [Google Scholar] [CrossRef]
- Poulsen, T.B.G.; Damgaard, D.; Jorgensen, M.M.; Senolt, L.; Blackburn, J.M.; Nielsen, C.H.; Stensballe, A. Identification of Novel Native Autoantigens in Rheumatoid Arthritis. Biomedicines 2020, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Ge, C.; Lonnblom, E.; Lin, X.; Feng, H.; Xiao, L.; Bai, J.; Ayoglu, B.; Nilsson, P.; Nandakumar, K.S.; et al. The autoantibody response to cyclic citrullinated collagen type II peptides in rheumatoid arthritis. Rheumatology 2019, 58, 1623–1633. [Google Scholar] [CrossRef]
- Sidney, J.; Vela, J.L.; Friedrich, D.; Kolla, R.; von Herrath, M.; Wesley, J.D.; Sette, A. Low HLA binding of diabetes-associated CD8+ T-cell epitopes is increased by post translational modifications. BMC Immunol. 2018, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Vomund, A.N.; Peterson, O.J.; Chervonsky, A.V.; Lichti, C.F.; Unanue, E.R. The MHC-II peptidome of pancreatic islets identifies key features of autoimmune peptides. Nat. Immunol. 2020, 21, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Olofsson, P.; Gelderman, K.A.; Holmberg, J.; Holmdahl, R. A new arthritis therapy with oxidative burst inducers. PLoS Med. 2006, 3, e348. [Google Scholar] [CrossRef]
- Hultqvist, M.; Backlund, J.; Bauer, K.; Gelderman, K.A.; Holmdahl, R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J. Immunol. 2007, 179, 1431–1437. [Google Scholar] [CrossRef]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef]
- Chiang, C.L.; Hagemann, A.R.; Leskowitz, R.; Mick, R.; Garrabrant, T.; Czerniecki, B.J.; Kandalaft, L.E.; Powell, D.J., Jr.; Coukos, G. Day-4 myeloid dendritic cells pulsed with whole tumor lysate are highly immunogenic and elicit potent anti-tumor responses. PLoS ONE 2011, 6, e28732. [Google Scholar] [CrossRef]
- Chiang, C.L.; Kandalaft, L.E.; Tanyi, J.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Chiang, C.L.; Tanyi, J.; Motz, G.; Balint, K.; Mick, R.; Coukos, G. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J. Transl. Med. 2013, 11, 149. [Google Scholar] [CrossRef]
- Martin Lluesma, S.; Wolfer, A.; Harari, A.; Kandalaft, L.E. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines 2016, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, A.; Graciotti, M.; Kandalaft, L.E.; Kandalaft, L. A cancer vaccine with dendritic cells differentiated with GM-CSF and IFNalpha and pulsed with a squaric acid treated cell lysate improves T cell priming and tumor growth control in a mouse model. Bioimpacts 2018, 8, 211–221. [Google Scholar] [CrossRef]
- Ophir, E.; Bobisse, S.; Coukos, G.; Harari, A.; Kandalaft, L.E. Personalized approaches to active immunotherapy in cancer. Biochim. Biophys. Acta 2016, 1865, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Powell, D.J., Jr.; Singh, N.; Coukos, G. Immunotherapy for ovarian cancer: What’s next? J. Clin. Oncol. 2011, 29, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Singh, N.; Liao, J.B.; Facciabene, A.; Berek, J.S.; Powell, D.J., Jr.; Coukos, G. The emergence of immunomodulation: Combinatorial immunochemotherapy opportunities for the next decade. Gynecol. Oncol. 2010, 116, 222–233. [Google Scholar] [CrossRef]
- Lin, A.G.; Xiang, B.; Merlino, D.J.; Baybutt, T.R.; Sahu, J.; Fridman, A.; Snook, A.E.; Miller, V. Non-thermal plasma induces immunogenic cell death in vivo in murine CT26 colorectal tumors. Oncoimmunology 2018, 7, e1484978. [Google Scholar] [CrossRef]
- Bekeschus, S.; Clemen, R.; Niessner, F.; Sagwal, S.K.; Freund, E.; Schmidt, A. Medical Gas Plasma Jet Technology Targets Murine Melanoma in an Immunogenic Fashion. Adv. Sci. 2020, 7, 1903438. [Google Scholar] [CrossRef]
- Lin, A.; Gorbanev, Y.; De Backer, J.; Van Loenhout, J.; Van Boxem, W.; Lemiere, F.; Cos, P.; Dewilde, S.; Smits, E.; Bogaerts, A. Non-Thermal Plasma as a Unique Delivery System of Short-Lived Reactive Oxygen and Nitrogen Species for Immunogenic Cell Death in Melanoma Cells. Adv. Sci. 2019, 6, 1802062. [Google Scholar] [CrossRef]
- Enomoto, K.; Sho, M.; Wakatsuki, K.; Takayama, T.; Matsumoto, S.; Nakamura, S.; Akahori, T.; Tanaka, T.; Migita, K.; Ito, M.; et al. Prognostic importance of tumour-infiltrating memory T cells in oesophageal squamous cell carcinoma. Clin. Exp. Immunol. 2012, 168, 186–191. [Google Scholar] [CrossRef]
- Koelzer, V.H.; Lugli, A.; Dawson, H.; Hadrich, M.; Berger, M.D.; Borner, M.; Mallaev, M.; Galvan, J.A.; Amsler, J.; Schnuriger, B.; et al. CD8/CD45RO T-cell infiltration in endoscopic biopsies of colorectal cancer predicts nodal metastasis and survival. J. Transl. Med. 2014, 12, 81. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Nigro, A.; Mentucci, F.M.; Rumie Vittar, N.B.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 12, 256. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Koks, C.; Verschuere, T.; Boon, L.; Van Gool, S.W.; Agostinis, P. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 2016, 8, 328ra27. [Google Scholar] [CrossRef] [PubMed]
- Vassilaros, S.; Tsibanis, A.; Tsikkinis, A.; Pietersz, G.A.; McKenzie, I.F.; Apostolopoulos, V. Up to 15-year clinical follow-up of a pilot Phase III immunotherapy study in stage II breast cancer patients using oxidized mannan-MUC1. Immunotherapy 2013, 5, 1177–1182. [Google Scholar] [CrossRef]
- Zhang, L.-X.; Sun, X.-M.; Jia, Y.-B.; Liu, X.-G.; Dong, M.; Xu, Z.P.; Liu, R.-T. Nanovaccine’s rapid induction of anti-tumor immunity significantly improves malignant cancer immunotherapy. Nano Today 2020, 35. [Google Scholar] [CrossRef]
- Stone, J.D.; Chervin, A.S.; Kranz, D.M. T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. [Google Scholar] [CrossRef]
- Baumgartner, C.K.; Yagita, H.; Malherbe, L.P. A TCR affinity threshold regulates memory CD4 T cell differentiation following vaccination. J. Immunol. 2012, 189, 2309–2317. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).