
The Evolution of Cancer Immunotherapy


Abstract
:1. Introduction
2. Cytokine Therapy
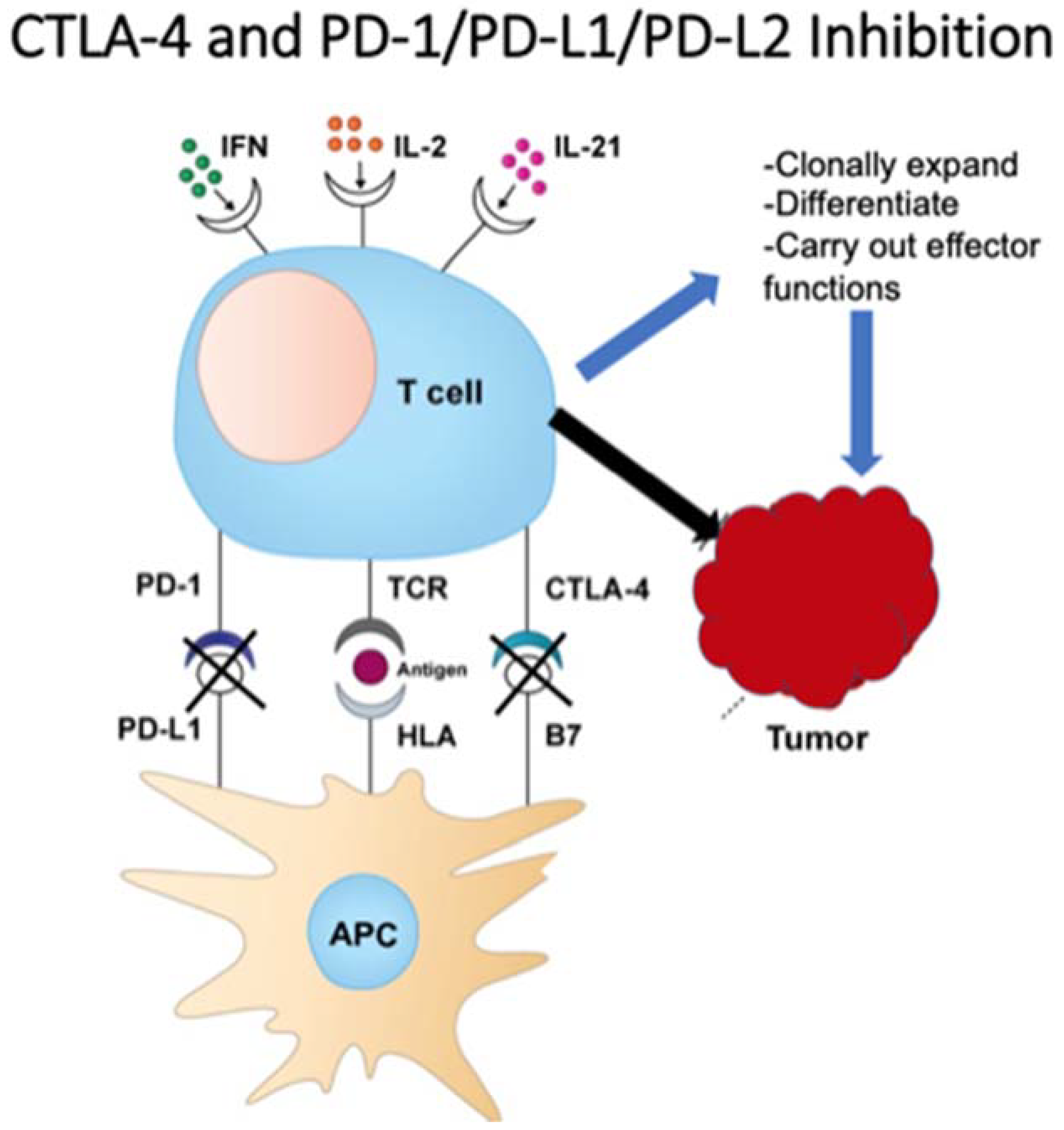
3. Checkpoint Inhibition
4. Antitumor Monoclonal Antibodies
5. CAR-T Cell Therapy
Bispecific Antibodies/BiTEs
6. Vaccines
6.1. Preventative Vaccines
6.2. Therapeutic Vaccines
7. Oncolytic Viruses
7.1. Neoantigen Vaccines
7.2. Combination Vaccine Therapies
7.3. Future Studies in Vaccines
8. Future/In Process
9. Conclusions
Funding
Conflicts of Interest
References
- Ichim, C.V. Revisiting immunosurveillance and immunostimulation: Implications for cancer immunotherapy. J. Transl. Med. 2005, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Coley, W.B. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar]
- Burnet, M. Cancer—A Biological Approach: III. Viruses Associated with Neoplastic Conditions. IV. Practical Applications. BMJ 1957, 1, 841–847. [Google Scholar] [CrossRef]
- Thomas, L.; Lawrence, H.S. Cellular and Humoral Aspects of the Hypersensitive States; Hoeber-Harper: New York, NY, USA, 1959. [Google Scholar]
- Teng, M.W.L.; Kershaw, M.H.; Smyth, M.J. Chapter 7—Cancer Immunoediting: From Surveillance to Escape. In Cancer Immunotherapy, 2nd ed.; Prendergast, G.C., Jaffee, E.M., Eds.; Academic Press: New York, NY, USA, 2013; pp. 85–99. ISBN 9780123942968. [Google Scholar]
- Houghton, A.N.; Guevara-Patiño, J.A. Immune recognition of self in immunity against cancer. J. Clin. Investig. 2004, 114, 468–471. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. OncoImmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [Green Version]
- Osipov, A.; Murphy, A.; Zheng, L. From immune checkpoints to vaccines: The past, present and future of cancer immunotherapy. Adv. Cancer Res. 2019, 143, 63–144. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.A.; Ruscetti, F.W.; Gallo, R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 1976, 193, 1007–1008. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Trotta, E.; Simeonov, D.R.; Marson, A.; Bluestone, J.A. Revisiting IL-2: Biology and therapeutic prospects. Sci. Immunol. 2018, 3, eaat1482. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Matsui, H.; Fujita, T.; Takaoka, C.; Kashima, N.; Yoshimoto, R.; Hamuro, J. Structure and expression of a cloned cDNA for human interleukin-2. Nat. Cell Biol. 1983, 302, 305–310. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Mulé, J.J.; Spiess, P.J.; Reichert, C.M.; Schwarz, S.L. Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J. Exp. Med. 1985, 161, 1169–1188. [Google Scholar] [CrossRef] [PubMed]
- Klapper, J.A.; Downey, S.G.; Smith, F.O.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Sherry, R.M.; Royal, R.E.; Steinberg, S.M.; Rosenberg, S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma. Cancer 2008, 113, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Dutcher, J.P.; Schwartzentruber, D.J.; Kaufman, H.L.; Agarwala, S.S.; Tarhini, A.A.; Lowder, J.N.; Atkins, M.B. High dose interleukin-2 (Aldesleukin)-expert consensus on best management practices-2014. J. Immunother. Cancer 2014, 2, 26. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Chambers, C.A.; Kuhns, M.S. CTLA-4-Mediated Inhibition in Regulation of T Cell Responses: Mechanisms and Manipulation in Tumor Immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. FDA Approves New Treatment for a Type of Late-Stage Skin Cancer. Available online: https://www.prnewswire.com/news-releases/fda-approves-new-treatment-for-a-type-of-late-stage-skin-cancer-118656794.html (accessed on 5 March 2021).
- Tarhini, A.; Lo, E.; Minor, D.R. Releasing the Brake on the Immune System: Ipilimumab in Melanoma and Other Tumors. Cancer Biother. Radiopharm. 2010, 25, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- George, A.P.; Kuzel, T.M.; Zhang, Y.; Zhang, B. The Discovery of Biomarkers in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 484–497. [Google Scholar] [CrossRef]
- Keegan, P. Center for Drug Evaluation and Research Summary Review-125514Orig1s000. September 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125514Orig1s000PharmR.pdf (accessed on 11 May 2021).
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Puri, S.; Shafique, M. Combination checkpoint inhibitors for treatment of non-small-cell lung cancer: An update on dual anti-CTLA-4 and anti-PD-1/PD-L1 therapies. Drugs Context 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- PD-1/PD-L1 Landscape. Cancer Research Institute. Available online: https://www.cancerresearch.org/scientists/immuno-oncology-landscape/pd-1-pd-l1-landscape (accessed on 5 March 2021).
- Zhao, J.; Chen, Y.; Ding, Z.-Y.; Liu, J.-Y. Safety and Efficacy of Therapeutic Cancer Vaccines Alone or in Combination with Immune Checkpoint Inhibitors in Cancer Treatment. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nat. Cell Biol. 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Pierpont, T.M.; Limper, C.B.; Richards, K.L. Past, Present, and Future of Rituximab—The World’s First Oncology Monoclonal Antibody Therapy. Front. Oncol. 2018, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C. HER2 Inhibition: From Discovery to Clinical Practice. Clin. Cancer Res. 2007, 13, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Vu, T.T.; Claret, F.X. Trastuzumab: Updated Mechanisms of Action and Resistance in Breast Cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Wang, Y.; Sun, T.; Wan, D.; Sheng, L.; Li, W.; Zhu, H.; Li, Y.; Lu, J. Overall Survival Benefit from Trastuzumab-Based Treatment in HER2-Positive Metastatic Breast Cancer: A Retrospective Analysis. Oncol. Res. Treat. 2018, 41, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Cruz, E.; Kayser, V. Monoclonal antibody therapy of solid tumors: Clinical limitations and novel strategies to enhance treatment efficacy. Biol. Targets Ther. 2019, 13, 33–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oostra, D.R.; Macrae, E.R. Role of trastuzumab emtansine in the treatment of HER2-positive breast cancer. Breast Cancer Targets Ther. 2014, 6, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z. From the Mouse Cage to Human Therapy: A Personal Perspective of the Emergence of T-bodies/Chimeric Antigen Receptor T Cells. Hum. Gene Ther. 2014, 25, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Sermer, D.; Brentjens, R. CAR T-cell therapy: Full speed ahead. Hematol. Oncol. 2019, 37, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Seimetz, D.; Heller, K.; Richter, J. Approval of First CAR-Ts: Have We Solved All Hurdles for ATMPs? Cell Med. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Munisvaradass, R.; Kumar, S.; Govindasamy, C.; Alnumair, K.S.; Mok, P.L. Human CD3+ T-Cells with the Anti-ERBB2 Chimeric Antigen Receptor Exhibit Efficient Targeting and Induce Apoptosis in ERBB2 Overexpressing Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18, 1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weimin, S.; Abula, A.; Qianghong, D.; Wenguang, W. Chimeric cytokine receptor enhancing PSMA-CAR-T cell-mediated prostate cancer regression. Cancer Biol. Ther. 2020, 21, 570–580. [Google Scholar] [CrossRef]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Dev. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M. Tumour virus vaccines: Hepatitis B virus and human papillomavirus. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160268. [Google Scholar] [CrossRef] [Green Version]
- Guallar-Garrido, S.; Julián, E. Bacillus Calmette-Guérin (BCG) Therapy for Bladder Cancer: An Update. Immunotargets Ther. 2020, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Alhunaidi, O.; Zlotta, A.R. The use of intravesical BCG in urothelial carcinoma of the bladder. Ecancermedicalscience 2019, 13, 905. [Google Scholar] [CrossRef] [Green Version]
- Amgen Inc. Imlygic™ (Talimogene Laherparepvec) Suspension for Intralesional Injection: US Prescribing Information. 2015. Available online: http://www.fda.gov/biologicsbloodvaccines (accessed on 1 February 2021).
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Dermime, S.; Armstrong, A.; Hawkins, R.E.; Stern, P.L. Cancer vaccines and immunotherapy. Br. Med. Bull. 2002, 62, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Lopes, A.; Vandermeulen, G.; Préat, V. Cancer DNA vaccines: Current preclinical and clinical developments and future perspectives. J. Exp. Clin. Cancer Res. 2019, 38, 146. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Dummer, R.; Ribas, A.; Puzanov, I.; Michielin, O.; Van der Walde, A.; Andtbacka, R.H.; Cebon, J.; Fernandez, E.; Malvehy, J.; et al. A Phase I/III, multicenter, open-label trial of talimogene laherparepvec (T-VEC) in combination with pembrolizumab for the treatment of unresected, stage IIIb-IV melanoma (MASTERKEY-265). J. Immunother. Cancer 2015, 3, P181. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.; Taylor, T.; Picconi, D.; Duma, C.; Aiken, R.; LaRocca, R.; Xiao-Tang, K.; Fu, B.; Alsharif, M.; Hsieh, C.; et al. ATIM-28. Phase II Trial of AV-GBM-1 (Autologous Dendritic Cells Loaded with Tumor Associated Antigens) as Adjunctive Therapy Following Surgery Plus Concurrent Chemoradiation in Newly Diagnosed GBM Patients. Neuro Oncol. 2019, 21 (Suppl. 6), vi7. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.-W. The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, J.; Wang, L.; Liu, B. Personalized neoantigen vaccination with synthetic long peptides: Recent advances and future perspectives. Theranostics 2020, 10, 6011–6023. [Google Scholar] [CrossRef] [PubMed]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations with Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Mao, Q.; Xia, W.; Dong, G.; Yu, C.; Jiang, F. Gut Microbiota Shapes the Efficiency of Cancer Therapy. Front. Microbiol. 2019, 10, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Microbiome Immunotherapy Toxicity and Response Evaluation—Full Text View. Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04107168 (accessed on 5 March 2021).
- Stern, L.A.; Jonsson, V.D.; Priceman, S.J. CAR T Cell Therapy Progress and Challenges for Solid Tumors. Cancer Treat. Res. 2020, 180, 297–326. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: Refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Drug Name | Target | Malignancy Approved (Single Agent or in Combination) | Approval Year |
|---|---|---|---|
| Atezolizumab | PD-L1 | Multiple solid tumors | 2016 |
| Bevacizumab | VEGF-A | Multiple solid tumors | 2004 |
| Brentuximab vedotin | CD30 | Hodgkin’s lymphoma; Anaplastic LCL; PTCL | 2011 |
| Blinatumomab | CD3, CD19 | Acute lymphoblastic leukemia | 2014 |
| Cemiplimab | PD-1 | Cutaneous squamous cell carcinoma | 2018 |
| Cetuximab | EGFR | Colorectal cancer (K-RAS wildtype); NSCLC, SCC head and neck | 2004 |
| Daratumumab | CD38 | Multiple myeloma | 2015 |
| Durvalumab | PD-L1 | Urothelial carcinoma, NSCLC, small cell lung cancer | 2017 |
| Elotuzumab | SLAMF7 | Multiple myeloma | 2015 |
| Gemtuzumab ozogamicin | CD33 | Acute myeloid leukemia | 2000 |
| Ipilimumab | CTLA-4 | Multiple solid tumors | 2011 |
| Isatuximab | CD38 | Multiple myeloma | 2020 |
| Mogamilizumab | CCR4 | Mycosis fungoides or Sezary syndrome, CTCL, T cell leukemia/lymphoma | 2018 |
| Nivolumab | PD-1 | Multiple solid tumors | 2014 |
| Obinutuzumab | CD20 | Chronic lymphocytic leukemia, follicular lymphoma | 2013 |
| Panitumumab | EGFR | Colorectal cancer | 2006 |
| Pembrolizumab | PD-1 | Multiple solid tumors | 2014 |
| Pertuzumab | HER2 | Breast cancer (HER2+) | 2012 |
| Ramucirumab | VEGFR2 | Multiple solid tumors | 2014 |
| Rituximab | CD20 | Multiple hematologic malignancies and autoimmune diseases | 1997 |
| Trastuzumab | HER2 | Breast cancer (HER2+), gastric/GEJ adenocarcinoma (HER2+) | 1998 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.; Maker, A.V.; Jain, S. The Evolution of Cancer Immunotherapy. Vaccines 2021, 9, 614. https://doi.org/10.3390/vaccines9060614
Khan M, Maker AV, Jain S. The Evolution of Cancer Immunotherapy. Vaccines. 2021; 9(6):614. https://doi.org/10.3390/vaccines9060614
Chicago/Turabian StyleKhan, Meshaal, Ajay V. Maker, and Shikha Jain. 2021. "The Evolution of Cancer Immunotherapy" Vaccines 9, no. 6: 614. https://doi.org/10.3390/vaccines9060614
APA StyleKhan, M., Maker, A. V., & Jain, S. (2021). The Evolution of Cancer Immunotherapy. Vaccines, 9(6), 614. https://doi.org/10.3390/vaccines9060614