3.1. Codon Optimization for Translation Elongation Efficiency
There are two levels of codon optimization. The first involves compound codon families. For example, SARS-2-S in the reference genome (NC_045512) contains 42 Arg residues, of which 30 are encoded by AGR codons and only 12 are encoded by CGN codons (S
Ref column in
Table 1). This avoidance of CGN codons makes evolutionary sense given that the host zinc finger antiviral proteins (ZAP, gene name
ZC3HAV1) target CpG dinucleotides in viral RNA and recruit cellular RNA degradation complexes to degrade the viral RNA genome [
27,
28,
29]. However, human genes use CGN more frequently than AGR codons for encoding Arg. Among the ribosomal protein genes (33 RPS and 50 RPL) known to be highly expressed, 64.2% of the Arg residues are encoded by CGN codons. BNT-162b2 and mRNA-1273 reduced AGR codons by 8 and 28, respectively, with the corresponding increase in CGN codons.
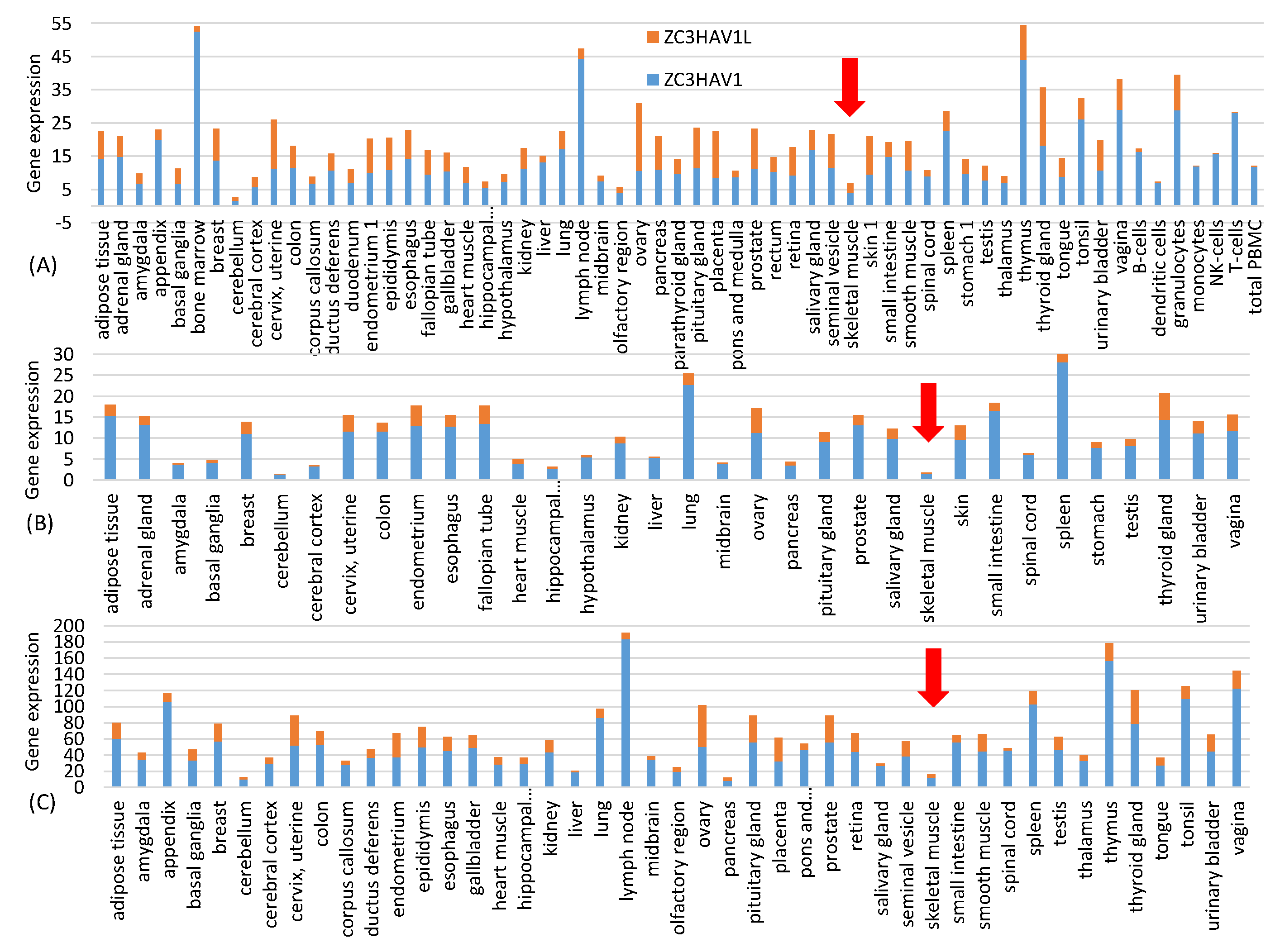
One might ask if the resulting increase in CpG dinucleotides would result in rapid degradation of the vaccine mRNA after being delivered into the host cell through the ZAP-mediated RNA degradation pathway [
27,
28,
29]. This is not a concern with the intramuscular injection because, according to three sets of gene expression data from Human Protein Atlas at
http://www.proteinatlas.org (accessed on 20 June 2021) [
30], ZAP is almost absent in skeletal muscle (
Figure 1). This highlights one advantage of mRNA vaccines because it has many different but convenient routes for vaccine administration, including subcutaneous, intramuscular, intradermal, intratracheal, intravenous and intraperitoneal routes [
31]. The high CpG in the vaccine mRNA provides two additional benefits. First, GC-rich mRNAs tend to be more stable than AU-rich mRNAs [
32]. Second, in the unlikely case when the vaccine mRNAs were recombined into a SARS-CoV-2 virus, the result would not be a virus with an optimized spike protein gene, but a segment of CpG-rich RNA that would be targeted by host ZAP for degradation.
The compound codon family for Leu is optimized similarly. Highly expressed human ribosomal protein genes encode 81% of Leu by CUN codons. For this reason, almost all UUR codons for Leu were recoded to CUN codons in both vaccine mRNAs (
Table 1). The compound codon family for Ser introduces a new twist. Both codon subfamilies are used roughly equally for encoding Ser. However, it is easier to optimize the AGY subfamily because AGC is clearly the preferred codon over AGU. Note that mutation bias would have favored U-ending codons because the frequency of U in introns is higher than that of C (0.2920 for U and 0.2178 for C,
Table 1), but highly expressed ribosome protein genes prefer AGC over AGU (144 for AGC and 95 for AGU,
Table 1). In contrast, the UCN subfamily has both UCC and UCU used frequently. For this reason, many Ser codons UCN were recoded to AGC in the vaccine mRNAs, especially in mRNA-1273 (
Table 1).
The second level of codon optimization is within-family optimization. Two strategies have been used. The first, referred to hereafter as the fundamentalist strategy, is simply to replace all codons by the major codon. Which codon is a major codon depends conceptually on two criteria: (1) the codon is preferred by highly expressed genes, and (2) it is decoded by the most abundant tRNA. However, superficial application of these two criteria can lead to mistakes. I will take the CGN codon family for Arg to show an incorrect optimization of the two mRNA vaccines.
The designers of both vaccines considered CGG as the optimal codon in the CGN codon family and recoded almost all CGN codons to CGG. This choice of CGG as the optimal codon seemingly resulted from application of both criteria above. First, the EMBOSS [
33] compilation of codon usage, which is frequently used in codon optimization, shows that CGG is used slightly more frequently than CGC. Second, CGG seems to have more tRNA decoding it than other synonymous CGN codons. A human genome contains seven tRNA
Arg/ACG genes (where superscripted ACG is the anticodon, with A deaminated to inosine I) to decode CGY codons, four tRNA
Arg/CCG genes to decode CGG codons and six tRNA
Arg/UCG genes to decode CGA and CGG (through wobble pairing at third codon site). Assuming that tRNA abundance is well correlated with tRNA gene copy number, which is true for
Saccharomyces cerevisiae [
34] but not known for other eukaryotes, one can infer that CGG is translated by more tRNAs genes (four tRNA
Arg/CCG genes six tRNA
Arg/UCG genes) than other codons and therefore is the major codon based on the two criteria. The two vaccines recoded nearly all CGN codons to CGG (
Table 1).
The reasoning above involving tRNA gene copy number is problematic. Nearly half of human tRNA genes are not expressed [
35], so we cannot use tRNA gene copy number as a proxy of tRNA abundance in the cellular tRNA pool. For this reason, codon preference by highly expressed genes relative to lowly expressed genes is a better operational criterion for codon optimization. The codon compilation of human genes in EMBOSS [
33] was done in 1993 and 1994 and did not aim to include only the highly expressed, so the slightly higher usage of CGG than CGC may simply be due to mutation bias (The frequency for nucleotide G is consistently higher than that of C in human introns).
There are two lines of evidence suggesting that CGG is not the optimal codon. The first involves the codon usage of human ribosomal protein genes (“RP” in
Table 1) which are known to be highly expressed. These genes prefer CGC codons (
Table 1). The second and more direct evidence is from codon usage of genes highly expressed in skeletal muscle cells (which are relevant here because the vaccine mRNA is injected and carried by the lipid nanoparticles into skeletal muscle cells to be translated, although vaccine mRNA could also be carried to some other tissues). I chose 50 genes most highly expressed in skeletal muscles from the consensus expression data set in Human Protein Atlas at
http://www.proteinatlas.org (accessed on 20 June 2021) [
30], but excluded those with CDSs with fewer than 300 codons. The remaining 26 genes (
Table 2), including the most muscle-specific genes such as titin (
TTN), actin (
ACTA1) and myosin (
MYH1), use CGC codons significantly more than CGG codons (Paired sample
t-test,
t = 3.075, DF = 25,
p = 0.0034, 2-tailed test). Therefore, the CGC codon preferred by ribosomal protein genes are also preferred by highly expressed muscle genes. Other protein-coding genes that are highly expressed are the two isoforms of human elongation factor 1α (
hEF1A1 and
hEF1A2), and poly(A)-binding protein (
hPABPC1). They also use more CGC than CGG (CGC:CGG are 3:0 for
hEF1A1, 8:6 for
hEF1A2, and 14:4 for
hPABPC1). These multiple lines of evidence suggest that CGC is a better codon than CGG. The designers of the mRNA vaccines (especially mRNA-1273,
Table 1) chose a wrong codon as the optimal codon.
Optimization of other codon families are straightforward. For 2-fold R-ending codons, background mutation bias, as reflected by nucleotide frequencies of introns in human genome, favors A-ending codons, but ribosomal protein genes consistently favor G-ending codons in every 2-fold R-ending codon family. Consequently, G-ending codons were taken as the optimal codon in the two mRNA vaccines (
Table 3 for GAR codons encoding Glu). For 2-fold Y-ending codons, the background mutation favors U-ending codons, but ribosomal protein genes favor C-ending codons, so C-ending codon is the optimal codon. There is another reason for recoding U-ending codons to C-ending codons. All U nucleotides in the two mRNA vaccines were replaced by N1-methylpseudouridines (Ψ) which can wobble with all for nucleotides and, therefore, should not be used in 2-fold codon families. For example, GAΨ encoding Asp could pair with the anticodon of tRNA
Glu leading to nonsynonymous substitutions. C-ending codons do not have this problem, which serves as another reason for recoding U-ending codons to C-ending codons.
The second strategy in codon optimization, referred to hereafter as the liberal strategy, is simply a less extreme version of the fundamentalist strategy that replaces all synonymous codons by the optimal codon. Suppose a synonymous codon family NNR with NNG decoded by tRNA-1 and NNA decoded by tRNA-2. Additionally, suppose that tRNA-1 is twice as abundant as tRNA-2 and that highly expressed genes favor NNG codon over NNA codon. The fundamentalist strategy is to replace all codons by NNG. The liberal strategy is based on the following rationale. When a cell is full of mRNA with NNG codons, tRNA-1 will be under such a high demand that it may become less available than tRNA-2, although there are twice as many tRNA-1 in the cell than tRNA-2. For this reason, it might be more optimal to keep some codons decoded by tRNA-2.
These two strategies are exemplified by the codon optimization involving GAR codons encoding Glu (
Table 3). The SARS-CoV-2 reference genome (NC_045512) has 34 GAA codons and 14 GAG codons in its spike protein gene. Moderna’s mRNA-1273 has taken the fundamentalist strategy and replaced all GAA codons by GAG. In contrast, Pfizer/BioNTech’s BNT-162b2 took the liberal strategy, and left 14 GAA codons unchanged (
Table 3). Moderna has consistently applied the fundamentalist strategy for all codon families in mRNA-1273, whereas Pfizer/BioNTech has consistently used the liberal strategy in codon optimization for BNT-162b2. There is no systematic evaluation of these two codon optimization strategies in translation efficiency. Given the difference in dosage (100 μg with mRNA-1273 and 30 μg with BNT-162b2) and the equivalence in efficacy, one may assume that an injection of Pfizer/BioNTech or Moderna vaccine produces the same number of the encoded spike proteins. This would imply that mRNA in the Pfizer/BioNTech vaccine on average likely produces about 3.3 times as many proteins as an mRNA in the Moderna vaccine.
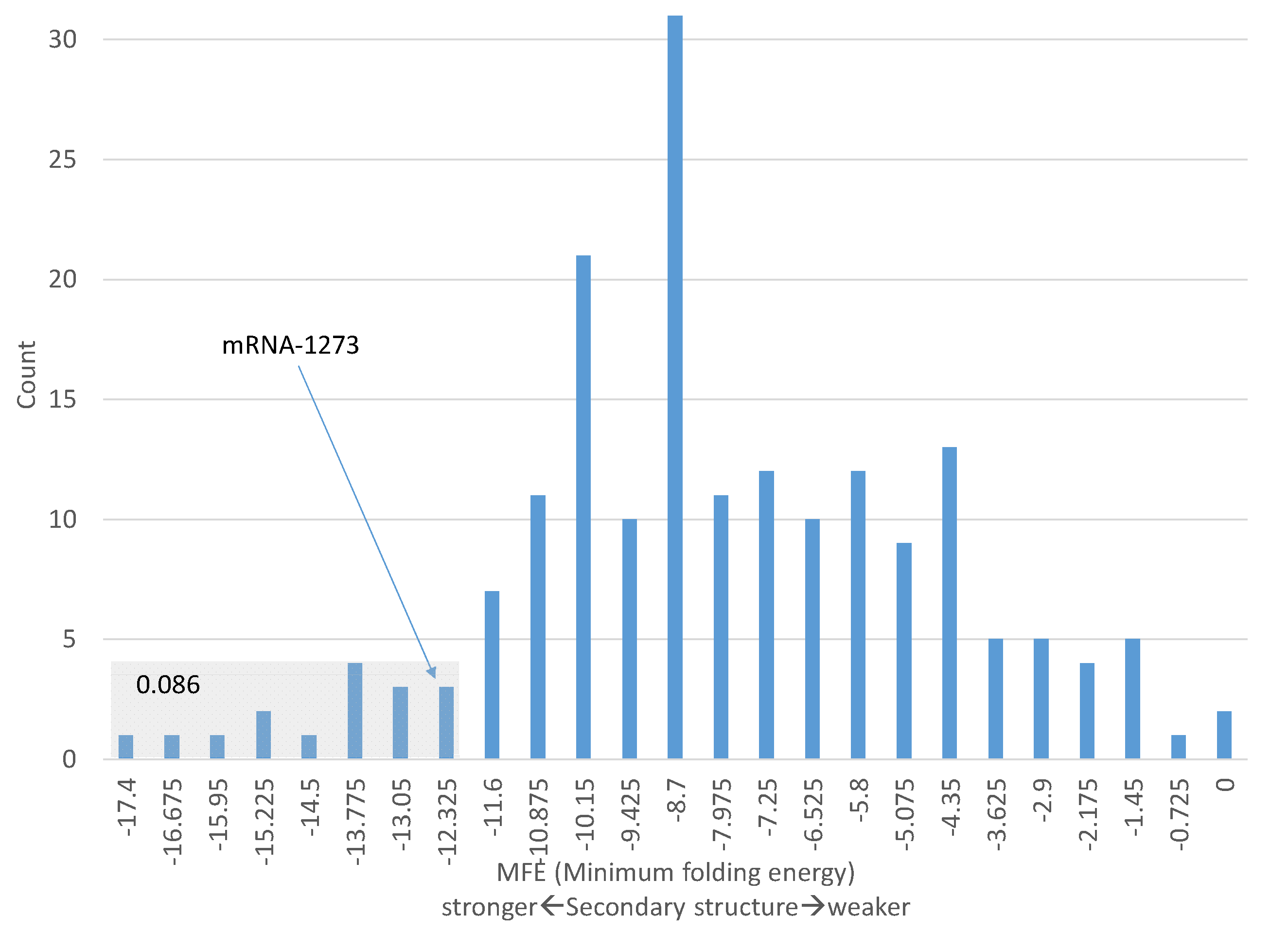
The codon optimization applied to BNT-162b2 and mRNA-1273 leads to a much increased codon adaptation index (CAI) [
21,
22] and index of translation efficiency (I
TE) [
11,
36] for the two vaccine mRNAs. The S gene from natural coronaviruses have CAI < 0.7 for their spike protein CDS, but the two codon-optimized spike CDSs have CAI equal to 0.94925 and 0.97939, respectively (
Table 4). I
TE is a generalized CAI taking into consideration of background mutation bias [
11]. Its values are similarly much higher in the two vaccine mRNAs than in natural viruses. The maximum CAI and I
TE values are 1.
The smaller value of CAI and ITE values for BNT-162b2 than mRNA-1273 might give an impression that BNT-162b2 is less codon-optimized than mRNA-1273. This is not necessarily true. As I mentioned before, mRNA-1273 was codon-optimized with the fundamentalist strategy (i.e., replacing all or almost all synonymous codons by the optimal codon), whereas BNT-162b2 was optimized with the liberal strategy which is less extreme than the first. The fundamentalist strategy will necessarily generate higher CAI or ITE values than the liberal strategy. However, the liberal strategy might lead to more efficient translation elongation if there are too many codons demanding the most abundant tRNA, as I discussed before.
3.2. Codon Optimization for Translation Accuracy
The codon optimization in the previous section suffers from the lack of consideration for translation accuracy [
36,
37]. Take Asn codons AAC and AAU in
E. coli for example. AAC is a major codon (heavily used by highly expressed genes and decoded by the most abundant isoacceptor tRNA) whereas AAU is a rarely used minor codon. Highly expressed
E. coli genes use AAC almost exclusively to encode Asn, so one could argue that the overuse of AAC is driven by selection for translation efficiency. However, AAC and AAU also differ in misreading rate, in particular by tRNA
Lys, which ideally should decode only AAA and AAG codons but does misread AAC and AAU, leading to Asn replaced by Lys. This misreading error rate is six times greater for AAU than for AAC, with the error ratio consistently maintained in different experimental settings, e.g., under both Asn-starved and non-starved conditions [
38], or with Streptomycin used to inhibit translation [
39]. Therefore, the overuse of AAC by highly expressed
E. coli genes could be driven either by selection for increased translation efficiency or increased translation accuracy or both.
Akashi [
37] attempted to disentangle the effect of selection on translation efficiency and accuracy. He classified amino acid sites into conserved sites (assumed to be functionally important) and variable sites (assumed to be of limited importance). If codon adaptation is due to selection for translation efficiency, then all codons in the gene should be subject to similar selection regardless of whether the codon is in a functionally important or unimportant site. In contrast, if codon adaptation is driven by selection for translation accuracy, then the selection is stronger in functionally important sites than in functionally unimportant sites. This implies greater select effect on functionally important codon sites than functionally unimportant codon sites. He found greater codon adaptation in conserved amino acid sites than in variable amino acid sites. This is consistent with his inference that the difference between the conserved and variable sites has resulted from selection for accuracy.
The observation, however, is also consistent with selection for translation efficiency. Take lysine codons (AAA and AAG) and glutamate codons (GAA and GAG) for example. Suppose that AAA codon can be decoded more efficiently than AAG, and GAG decoded more efficiently than GAA. Additionally, suppose that a highly expressed ancestral gene has evolved strong codon adaptation with lysine coded mainly by AAA and glutamate coded mainly by GAG. Now, some lysine sites might happen to experience nonsynonymous substitutions from AAA to GAA. These sites are now designated as variable (functionally unimportant) sites and are occupied by a minor codon GAA. This would result in an association between “poor codon adaptation” and variable (functionally unimportant) sites that has little to do with translation accuracy. Akashi [
37] discussed this problem but did not provide a definitive solution.
There are two approaches to optimize codon usage for accuracy. The first is to empirically characterize the decoding error rate for each synonymous codon in skeletal muscle cells, and to choose the codon with the lowest error rate. For mRNA to be translated in
E. coli, then recoding AAU to AAC would increase accuracy because AAC has a misreading error six times smaller than AAU. An alternative is again to follow the codon usage of functionally important and highly expressed genes, such as ribosomal proteins or highly expressed genes in skeletal muscle cells in
Table 2. It is important for vaccine mRNA to be translated accurately because misincorporation of the wrong amino acids would confuse our immune system in target recognition.
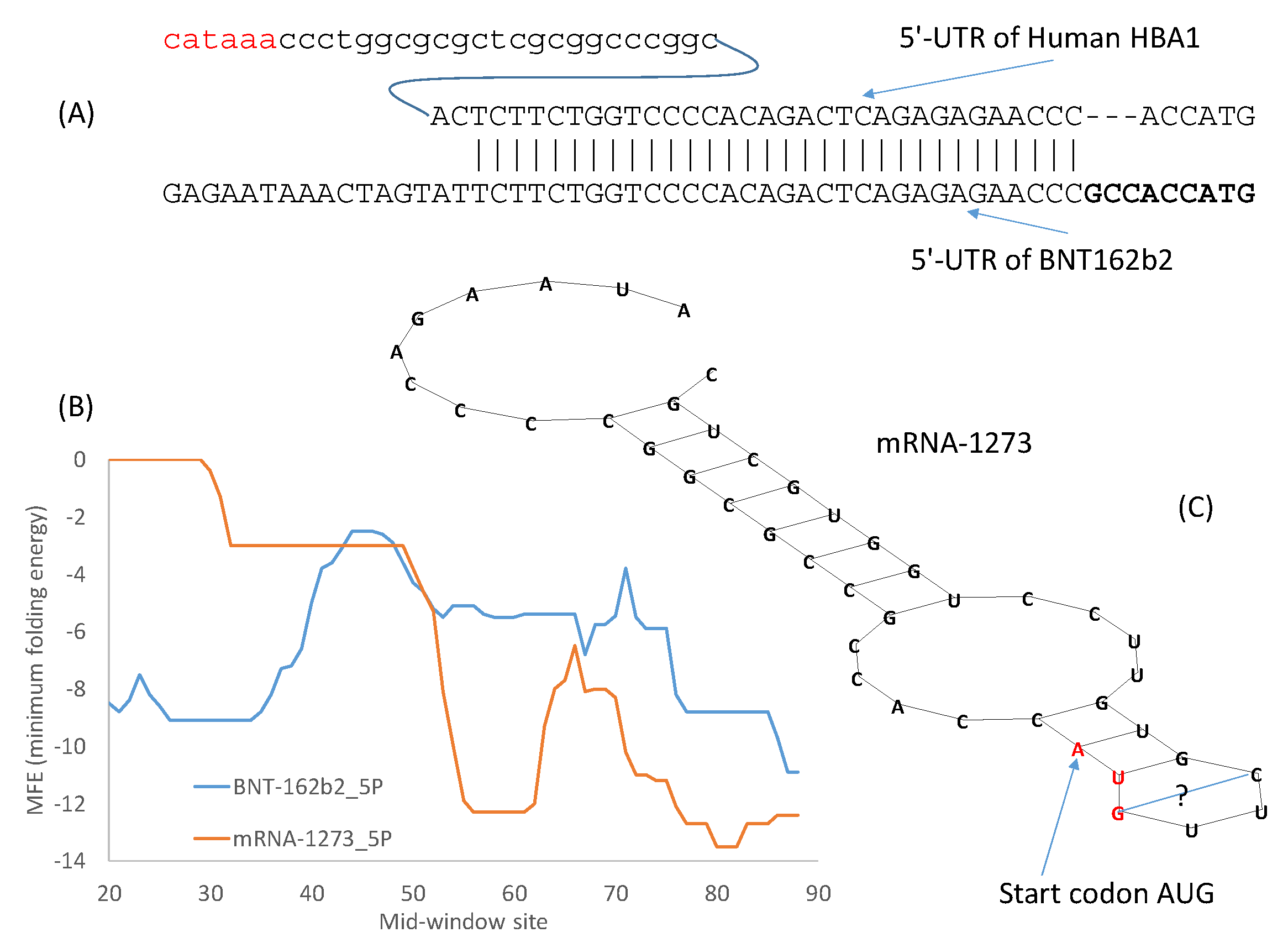
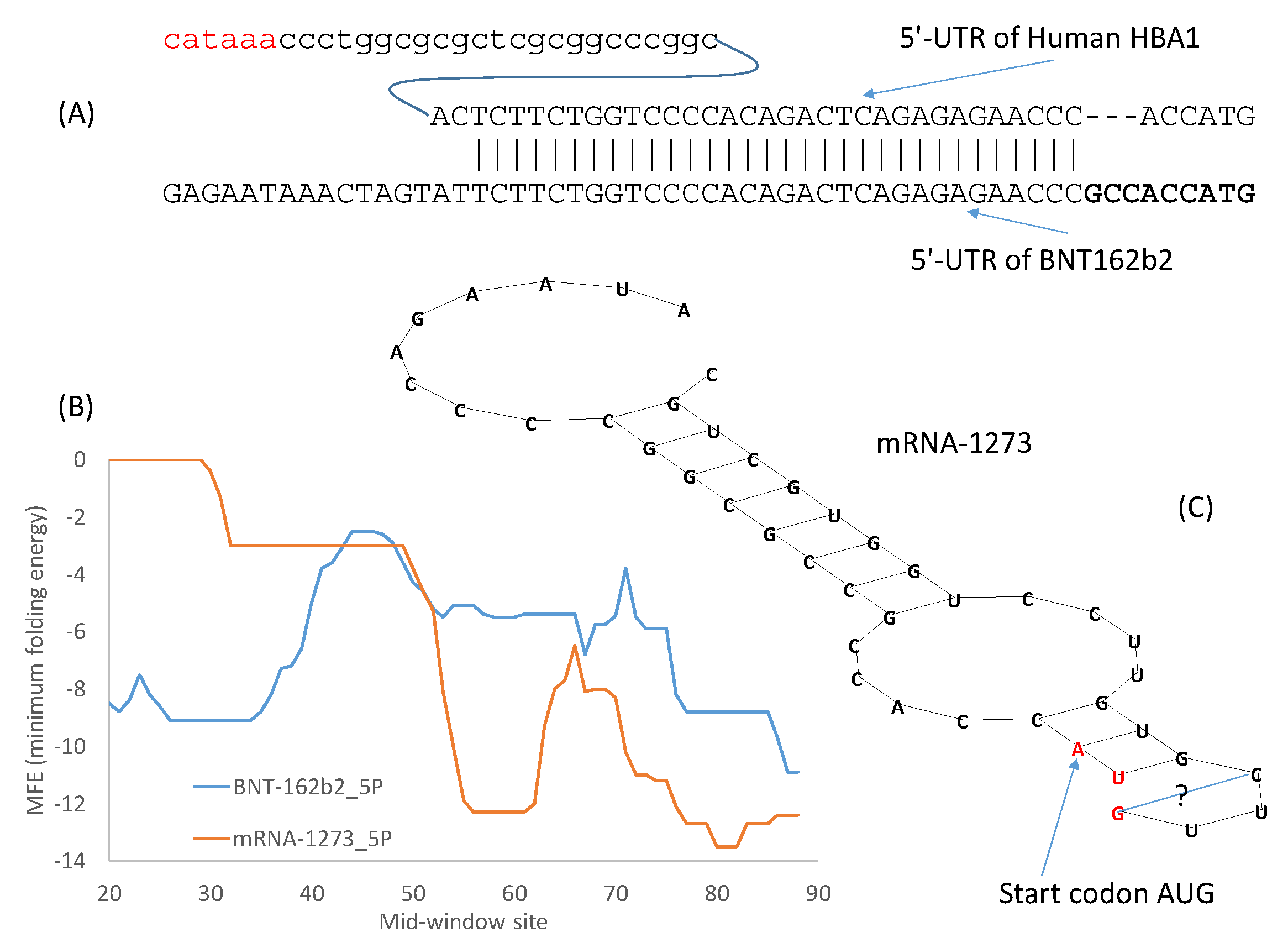
3.5. The 3′-UTR of mRNA Vaccines
I have previously mentioned different approaches for optimizing 5′-UTR and 3′-UTR. Given sufficient time, the systematic evolution of ligands by exponential enrichment (SELEX) [
54] should be the preferred method. However, in an emergency, the alternative approach of borrowing from nature could be more efficient. The 5′-UTR of the Pfizer/BioNTech vaccine mRNA incorporates the 5′-UTR of a human α-globin gene (
Figure 2A), which makes sense because α-globin mRNAs are translated very efficiently. The same approach of borrowing from nature has been used for designing 3′-UTR of therapeutic mRNAs, e.g., by incorporating stability regulatory elements from human α-globin and β-globin genes [
13]. These stability regulatory elements often form RNA-protein complexes to stabilize mRNA [
93,
94,
95,
96,
97]. The 5′-UTR and 3′-UTR of globin genes, when ligated to other mRNAs, can confer stability to these mRNAs [
54,
98,
99]. Moderna’s mRNA-1273 “pasted” the 110-nt 3′-UTR of human α-globin gene (
HBA1) between the last stop codon and a poly(A) tail.The design of the 3′-UTR of the Pfizer/BioNTech mRNA vaccine is a combination of SELEX and borrowing from nature. The objective is to find naturally occurring RNA segments that perform better than the 3′-UTR of human β-globin mRNA [
54]. Two RNA segments outperform other alternatives through the SELEX optimization protocol [
54]. One of them is from the human mitochondrial 12S rRNA (
mtRNR1), and the other segment is from human
AES/TLE5 gene. As these two RNA segments were found to have the lowest number of predicted binding sites for miRNAs and the highest hybridization energies [
54], two C→U mutations were introduced in the
AES segment to further increase the binding energy (from MFE = −37 to −39.3 at 37 °C, my calculation from DAMBE). For Pfizer/BioNTech’s mRNA vaccine, the
AES segment of 136 nt with the two C→Ψ mutations was pasted right after two trinucleotides following the second stop codon. The
mtRNR1 segment of 139 nt was pasted immediately after. This heuristic and empirical approach of borrowing from nature is perhaps more efficient than alternatives in an emergency.







{kind=link}
{kind=link}
{kind=link}
{kind=link}