
Towards an Ensemble Vaccine against the Pegivirus Using Computational Modelling Approaches and Its Validation through In Silico Cloning and Immune Simulation

 , ,
, ,  and
and 
Abstract
1. Introduction
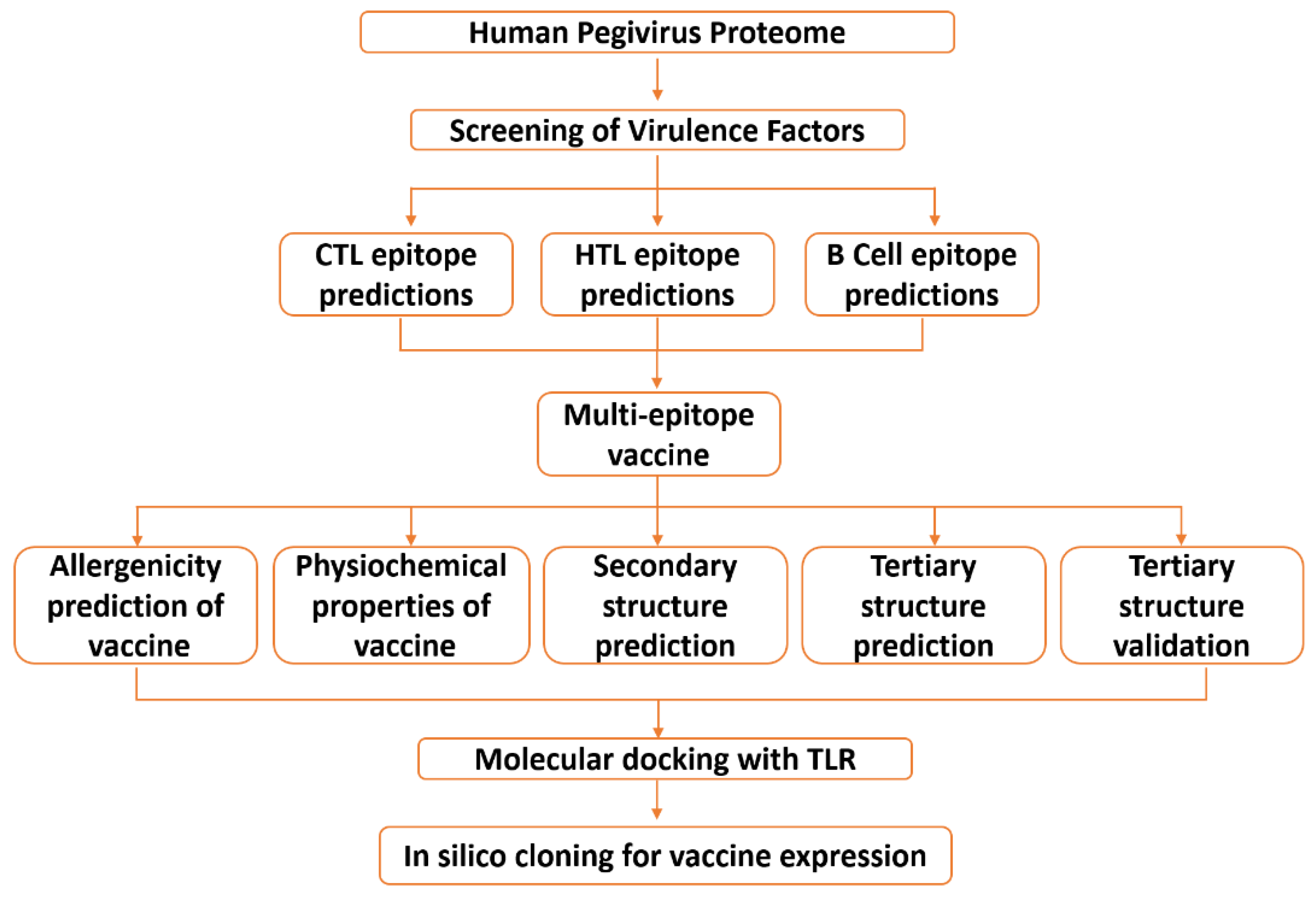
2. Materials and Methods
2.1. Data Retrieval
2.2. Data Processing
2.2.1. Mapping Immunogenic Peptides in the Pegivirus Proteome
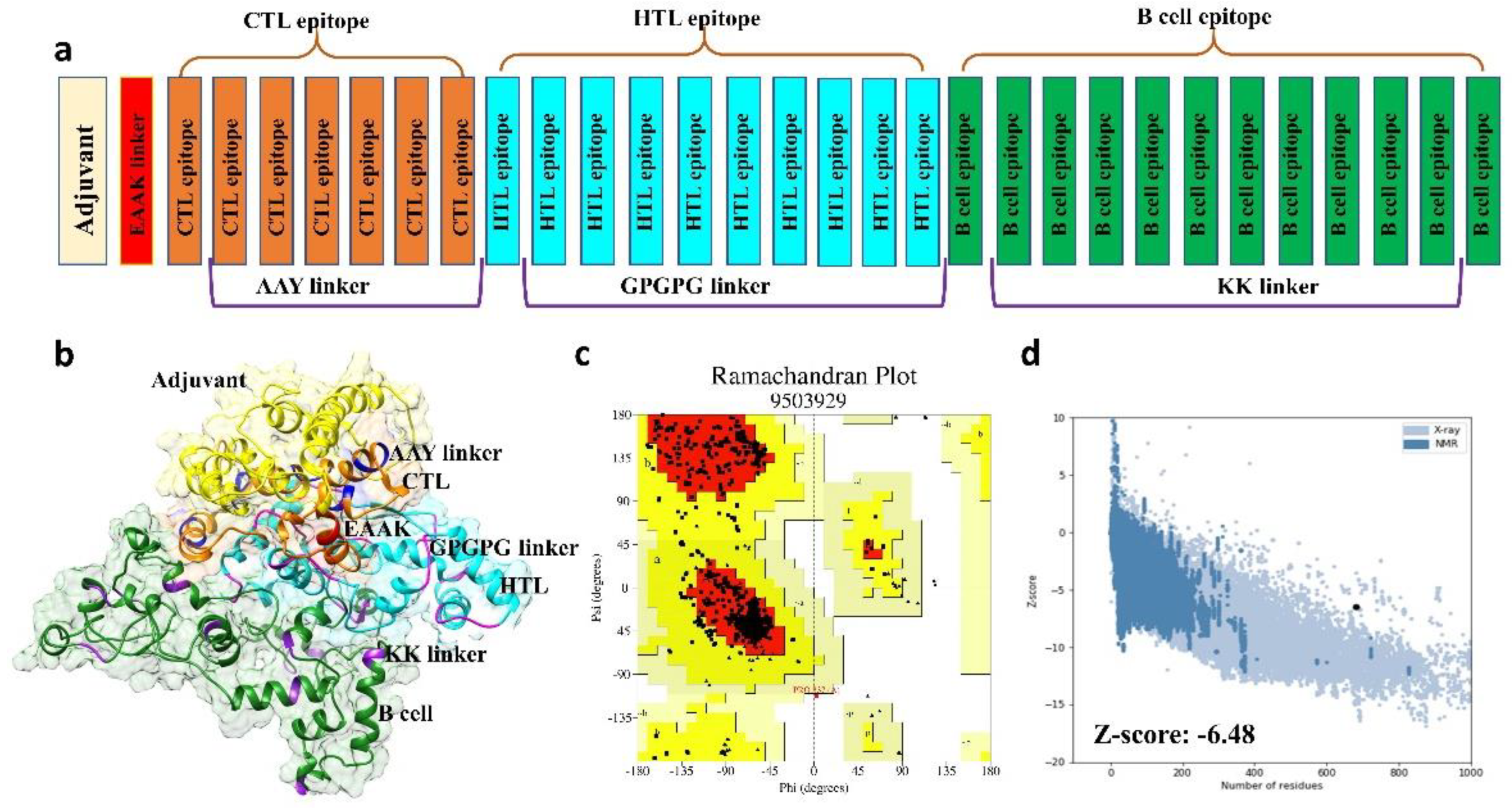
2.2.2. Multi-Epitope Vaccine Construction and Characterization
2.2.3. Structural Modeling, Refinement, and Validation of Vaccine Construct
2.3. Validation of the Vaccine Construct Immune Potential
2.3.1. Molecular Docking
2.3.2. Molecular Dynamic (MD) Simulation
2.3.3. In Silico Cloning and Optimization of Vaccine Protein
2.3.4. Immune Simulation
3. Results
3.1. Sequence Retrieval and Antigenicity Profiling
3.2. Immunogenic CTL, HTL, and B-Cell Epitopes Prediction
3.3. Structural Prediction of Final Vaccine Construct
3.4. 3D Structure Validation
3.5. Prediction of Allergenicity
3.6. Antigenicity of the Vaccine Construct
3.7. Physiochemical Parameters Prediction
3.8. Molecular Docking
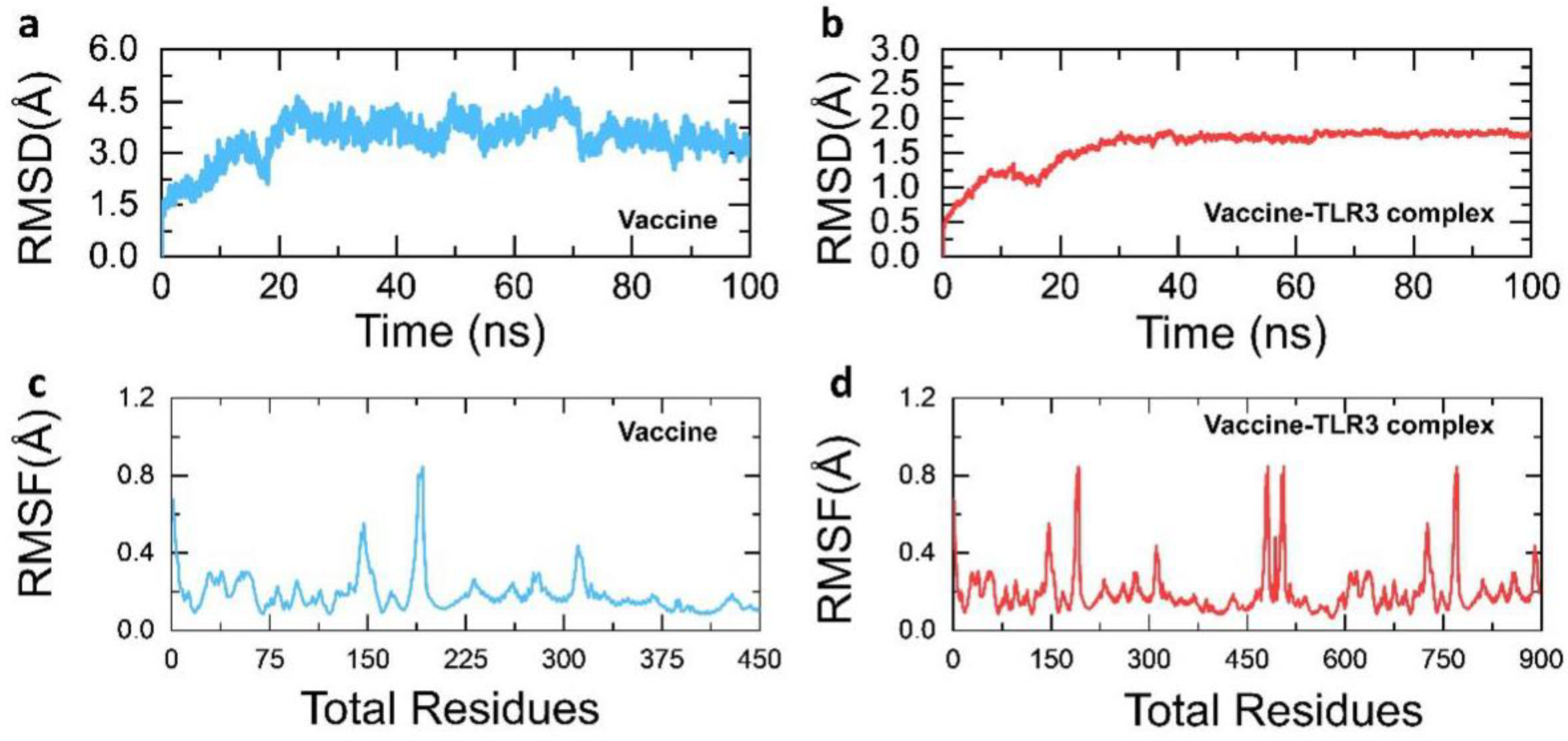
3.9. Structural Dynamics Features of the Vaccine Ensemble and TLR3 Complex
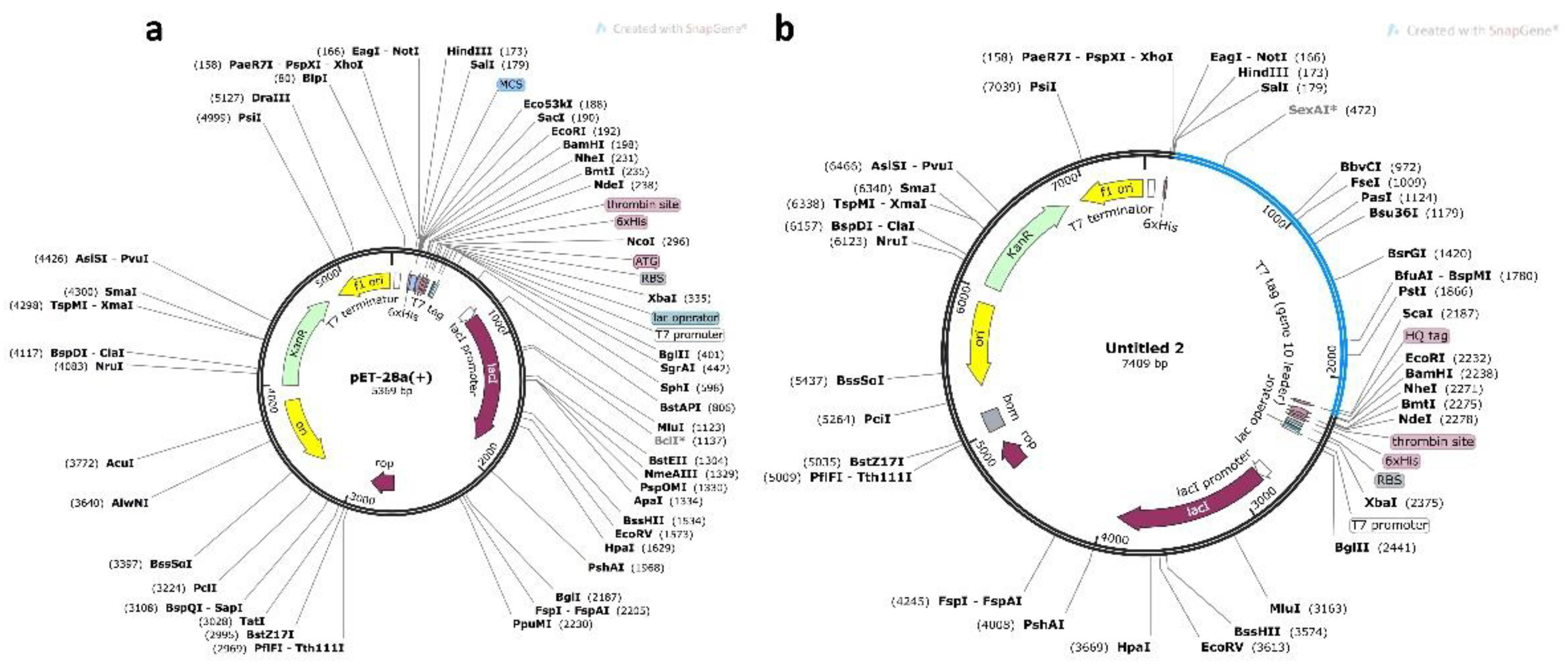
3.10. Codon Optimization and In Silico Cloning
3.11. Immune Simulation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arroyave-Ospina, J.C.; Caicedo, M.F.; Navas, M.C.; Cortes-Mancera, F.M. Human pegivirus: Pathogenic potential and non-hodgkin lymphoma development risk. Rev. Chil. Infectol. 2018, 35, 164–175. [Google Scholar] [CrossRef]
- Marano, G.; Franchini, M.; Farina, B.; Piccinini, V.; Pupella, S.; Vaglio, S.; Grazzini, G.; Liumbruno, G. The human pegivirus: A new name for an" ancient" virus. Can transfusion medicine come up with something new? Acta Virol. 2017, 61, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, J.T.; Foung, S.; Muerhoff, A.S.; Bukh, J.; Simmonds, P. The gb viruses: A review and proposed classification of gbv-a, gbv-c (hgv), and gbv-d in genus pegivirus within the family flaviviridae. J. Gen. Virol. 2011, 92, 233. [Google Scholar] [CrossRef]
- Vitrenko, Y.; Kostenko, I.; Kulebyakina, K.; Sorochynska, K. Prevalence of human pegivirus-1 and sequence variability of its e2 glycoprotein estimated from screening donors of fetal stem cell-containing material. Virol. J. 2017, 14, 1–11. [Google Scholar] [CrossRef]
- Polgreen, P.M.; Xiang, J.; Chang, Q.; Stapleton, J.T. Gb virus type c/hepatitis g virus: A non-pathogenic flavivirus associated with prolonged survival in hiv-infected individuals. Microbes Infect. 2003, 5, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhao, W.; Feng, Y.; Dai, J.; Li, Z.; Zhang, X.; Liu, L.; Bai, J.; Zhang, H.; Lu, L.; et al. A novel genotype of gb virus c: Its identification and predominance among injecting drug users in yunnan, china. PLoS ONE 2011, 6, e21151. [Google Scholar] [CrossRef]
- Horemheb-Rubio, G.; Ramos-Cervantes, P.; Arroyo-Figueroa, H.; Ávila-Ríos, S.; García-Morales, C.; Reyes-Terán, G.; Escobedo, G.; Estrada, G.; García-Iglesias, T.; Muñoz-Saucedo, N.; et al. High hpgv replication is associated with improved surrogate markers of hiv progression. PLoS ONE 2017, 12, e0184494. [Google Scholar] [CrossRef]
- Fama, A.; Larson, M.C.; Link, B.K.; Habermann, T.M.; Feldman, A.L.; Call, T.G.; Ansell, S.M.; Liebow, M.; Xiang, J.; Maurer, M.J.; et al. Human pegivirus infection and lymphoma risk: A systematic review and meta-analysis. Clin. Infect. Dis. 2020, 71, 1221–1228. [Google Scholar] [CrossRef]
- Chang, C.M.; Stapleton, J.T.; Klinzman, D.; McLinden, J.H.; Purdue, M.P.; Katki, H.A.; Engels, E.A. Gbv-c infection and risk of nhl among us adults. Cancer Res. 2014, 74, 5553–5560. [Google Scholar] [CrossRef]
- Zintzaras, E.; Voulgarelis, M.; Moutsopoulos, H.M. The risk of lymphoma development in autoimmune diseases: A meta-analysis. Arch. Intern. Med. 2005, 165, 2337–2344. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Sakata, K.; Okuzaki, D.; Inokuchi, S.; Tamura, T.; Motooka, D.; Nakamura, S.; Ono, C.; Shimokawa, M.; Matsuura, Y.; et al. Characterization of human pegivirus infection in liver transplantation recipients. J. Med. Virol. 2019, 91, 2093–2100. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, J.; Song, Y.; Wang, X.; Zhou, M.; Wang, L.; Ma, J.; Zhu, J. Mortality of lymphoma and myeloma in China, 2004–2017: An observational study. J. Hematol. Oncol. 2019, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Dwyer-Lindgren, L.; Fitzmaurice, C.; Stubbs, R.W.; Bertozzi-Villa, A.; Morozoff, C.; Charara, R.; Allen, C.; Naghavi, M.; Murray, C.J. Trends and patterns of disparities in cancer mortality among us counties, 1980–2014. JAMA 2017, 317, 388–406. [Google Scholar] [CrossRef]
- Lee, H.; Park, H.J.; Park, E.-H.; Ju, H.Y.; Oh, C.-M.; Kong, H.-J.; Jung, K.-W.; Park, B.-K.; Lee, E.; Eom, H.-S.; et al. Nationwide statistical analysis of lymphoid malignancies in Korea. Cancer Res. Treat. 2018, 50, 222. [Google Scholar] [CrossRef]
- Balcom, E.F.; Doan, M.A.; Branton, W.G.; Jovel, J.; Blevins, G.; Edguer, B.; Hobman, T.C.; Yacyshyn, E.; Emery, D.; Box, A.; et al. Human pegivirus-1 associated leukoencephalitis: Clinical and molecular features. Ann. Neurol. 2018, 84, 781–787. [Google Scholar] [CrossRef]
- Ansell, S.M.; Armitage, J. Non-Hodgkin Lymphoma: Diagnosis and Treatment; Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1087–1097. [Google Scholar]
- Hennessy, B.T.; Hanrahan, E.O.; Daly, P.A. Non-hodgkin lymphoma: An update. Lancet Oncol. 2004, 5, 341–353. [Google Scholar] [CrossRef]
- Salles, G.A. Clinical features, prognosis and treatment of follicular lymphoma. Hematology 2007, 2007, 216–225. [Google Scholar] [CrossRef][Green Version]
- Ansell, S.M. Non-Hodgkin Lymphoma: Diagnosis and Treatment; Mayo Clinic Proceedings; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1152–1163. [Google Scholar]
- WHO; UNICEF. Global Immunization Data; World Health Organization and UNICEF: Geneva, Switzerland; UNICEF: New York, NY, USA, 2007. [Google Scholar]
- Khan, A.; Khan, M.; Saleem, S.; Babar, Z.; Ali, A.; Khan, A.A.; Sardar, Z.; Hamayun, F.; Ali, S.S.; Wei, D.-Q. Phylogenetic analysis and structural perspectives of rna-dependent rna-polymerase inhibition from sars-cov-2 with natural products. Interdiscip. Sci. Comput. Life Sci. 2020, 12, 335–348. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.-Q. Higher infectivity of the sars-cov-2 new variants is associated with k417n/t, e484k, and n501y mutants: An insight from structural data. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef]
- Khan, A.; Khan, S.; Saleem, S.; Nizam-Uddin, N.; Mohammad, A.; Khan, T.; Ahmad, S.; Arshad, M.; Ali, S.S.; Suleman, M. Immunogenomics guided design of immunomodulatory multi-epitope subunit vaccine against the sars-cov-2 new variants, and its validation through in silico cloning and immune simulation. Comput. Biol. Med. 2021, 133, 104420. [Google Scholar] [CrossRef]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic t-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (iedb): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Khan, S.; Ali, A.; Akbar, H.; Sayaf, A.M.; Khan, A.; Wei, D.-Q. Immunoinformatics approaches to explore helicobacter pylori proteome (virulence factors) to design b and t cell multi-epitope subunit vaccine. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, L.; Venkatesan, A.; Febin Prabhu Dass, J. Epitope-based immunoinformatics approach on RNA-dependent RNA polymerase (RdRp) protein complex of nipah virus (NiV). J. Cell. Biochem. 2019, 120, 7082–7095. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Krutz, N.L.; Kern, P.S.; Gunalan, V.; Nguyen, M.N.; Limviphuvadh, V.; Eisenhaber, F.; Gerberick, G.F. Allercatpro—Prediction of protein allergenicity potential from the protein sequence. Bioinformatics 2019, 35, 3020–3027. [Google Scholar] [CrossRef]
- Zaharieva, N.; Dimitrov, I.; Flower, D.; Doytchinova, I. Immunogenicity prediction by vaxijen: A ten year overview. J. Proteom. Bioinform. 2017, 10, 298–310. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the expasy server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Laskowski, R.A.; Chistyakov, V.V.; Thornton, J.M. Pdbsum more: New summaries and analyses of the known 3d structures of proteins and nucleic acids. Nucleic Acids Res. 2005, 33, D266–D268. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Park, H.; Heo, L.; Seok, C. Galaxyweb server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. Prosa-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xia, M.; Chen, J.; Deng, F.; Yuan, R.; Zhang, X.; Shen, F. Data set for phylogenetic tree and rampage ramachandran plot analysis of sods in gossypium raimondii and g. Arboreum. Data Brief. 2016, 9, 345–348. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. Hdock: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham III, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. Amber, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. Jcat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Castiglione, F. Immune system simulation online. Bioinformatics 2011, 27, 2013–2014. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Oettgen, H.C. Adaptive immunity. J. Allergy Clin. Immunol. 2010, 125, S33–S40. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Keith Roberts, P.W. Molecular Biology of The Cell; Garland Science: New York, NY, USA, 2018. [Google Scholar]
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for t-and b-cell epitope prediction. J. Immunol. Res. 2017, 2017. [Google Scholar] [CrossRef]
- Chen, Z.; Ruan, P.; Wang, L.; Nie, X.; Ma, X.; Tan, Y. T and b cell epitope analysis of sars-cov-2 s protein based on immunoinformatics and experimental research. J. Cell. Mol. Med. 2020, 25, 1274–1289. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, A.J.; Maharaj, L.; Adeleke, V.T.; Okpeku, M.; Adeniyi, A.A.; Adeleke, M.A. Immunoinformatics prediction of overlapping cd8+ t-cell, ifn-γ and il-4 inducer cd4+ t-cell and linear b-cell epitopes based vaccines against covid-19 (sars-cov-2). Vaccine 2021, 39, 1111–1121. [Google Scholar] [CrossRef]
- Khan, A.; Junaid, M.; Kaushik, A.C.; Ali, A.; Ali, S.S.; Mehmood, A.; Wei, D.-Q. Computational identification, characterization and validation of potential antigenic peptide vaccines from hrhpvs e6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 2018, 13, e0196484. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Wei, D.-Q.; Kousar, K.; Abubaker, J.; Ahmad, S.; Ali, J.; Al-Mulla, F.; Ali, S.S.; Nizam-Uddin, N.; Sayaf, A.M.; et al. Preliminary structural data revealed that the sars-cov-2 b. 1.617 variant’s rbd binds to ace2 receptor stronger than the wild type to enhance the infectivity. ChemBioChem 2021. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Tworowski, D.; Detroja, R.; Mukherjee, S.B.; Frenkel-Morgenstern, M. Immunoinformatics and structural analysis for identification of immunodominant epitopes in sars-cov-2 as potential vaccine targets. Vaccines 2020, 8, 290. [Google Scholar] [CrossRef]
- Zheng, L.-N.; Lin, H.; Pawar, R.; Li, Z.-X.; Li, M.-H. Mapping ige binding epitopes of major shrimp (penaeus monodon) allergen with immunoinformatics tools. Food Chem. Toxicol. 2011, 49, 2954–2960. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue No | Peptide Sequence | MHC Binding Affinity | Rescale Binding Affinity | C-terminal Cleavage Affinity | Transport Affinity | Prediction Score | MHC-I Binding |
|---|---|---|---|---|---|---|---|
| 1511 | ATDALSTGY | 0.7930 | 3.3668 | 0.9637 | 2.9180 | 3.6573 | YES |
| 1690 | NSNKTPLLY | 0.6869 | 2.9163 | 0.9593 | 3.0490 | 3.2126 | YES |
| 126 | ATHPICWDY | 0.5329 | 2.2626 | 0.9680 | 3.2180 | 2.5687 | YES |
| 1442 | LTDTGDVEF | 0.5175 | 2.1971 | 0.9251 | 2.3890 | 2.4553 | YES |
| 1595 | AVESAMVFY | 0.5148 | 2.1858 | 0.4948 | 3.0140 | 2.4108 | YES |
| 2397 | YLTNKHSHY | 0.4236 | 1.7984 | 0.9649 | 2.9890 | 2.0925 | YES |
| 273 | HQSESYLKY | 0.4093 | 1.7378 | 0.9614 | 3.0060 | 2.0323 | YES |
| S. No | Allele | Start | End | Peptide Sequence | Method | Percentile Rank |
|---|---|---|---|---|---|---|
| 1 | HLA-DRB3*02:02 | 521 | 535 | ARRGFRMSNNPLSLL | NetMHCIIpan | 0.01 |
| 2 | HLA-DRB3*02:02 | 1970 | 1984 | LVFILSNSSVTTWAN | NetMHCIIpan | 0.01 |
| 3 | HLA-DRB1*07:01 | 3039 | 3053 | ASRLRFWLVASAILA | Consensus (comb.lib./smm/nn) | 0.02 |
| 4 | HLA-DRB3*02:02 | 1968 | 1982 | IVLVFILSNSSVTTW | NetMHCIIpan | 0.02 |
| 5 | HLA-DRB1*15:01 | 783 | 797 | AFLIYILSHPVNAAL | Consensus (smm/nn/sturniolo) | 0.13 |
| 6 | HLA-DRB5*01:01 | 757 | 771 | DGLFPIRHATAALRF | Consensus (smm/nn/sturniolo) | 0.13 |
| 7 | HLA-DRB5*01:01 | 1328 | 1342 | SVAVVKSMAPYIKET | Consensus (smm/nn/sturniolo) | 0.14 |
| 8 | HLA-DRB3*02:02 | 1202 | 1216 | SRVWVMNNNGGLVCG | NetMHCIIpan | 0.19 |
| 9 | HLA-DRB3*02:02 | 1683 | 1697 | KWKCLLNNSNKTPLL | NetMHCIIpan | 0.2 |
| 10 | HLA-DRB1*15:01 | 229 | 243 | GLLWQMFVSFPILYS | Consensus (smm/nn/sturniolo) | 0.21 |
| S. No | Position | Epitope | Score |
|---|---|---|---|
| 1 | 2494 | YNWFRSIVAPTTPPLPATRS | 1 |
| 2 | 1277 | SSSGGQGGMQAPAVTPTYSE | 1 |
| 3 | 494 | EQFGPGLGKWVPLPGEPVPE | 1 |
| 4 | 1340 | KETYKIRPEIRAGTGPDGVT | 1 |
| 5 | 1811 | DASRGASQYLAAAPPSPAPL | 1 |
| 6 | 2318 | VVQAASRFVPPVPKPRTRVS | 0.999 |
| 7 | 369 | NTTIIPQNCRNSTVDPTTAP | 0.999 |
| 8 | 1572 | GAYYTTSPGAAPCVSVPDAN | 0.999 |
| 9 | 663 | SCGHAVPPPDRGWEVPAAMS | 0.998 |
| 10 | 545 | APFCNPTPGRVRVCNNTAFY | 0.998 |
| 11 | 3000 | SPEVRTPQPEPKGMCLLPPE | 0.997 |
| 12 | 275 | SESYLKYCTITNTSTSMNCD | 0.996 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, B.; Suleman, M.; Zafar, Z.; Ali, S.S.; Nasir, S.N.; Namra; Hussain, Z.; Waseem, M.; Khan, A.; Hassan, F.; et al. Towards an Ensemble Vaccine against the Pegivirus Using Computational Modelling Approaches and Its Validation through In Silico Cloning and Immune Simulation. Vaccines 2021, 9, 818. https://doi.org/10.3390/vaccines9080818
Zheng B, Suleman M, Zafar Z, Ali SS, Nasir SN, Namra, Hussain Z, Waseem M, Khan A, Hassan F, et al. Towards an Ensemble Vaccine against the Pegivirus Using Computational Modelling Approaches and Its Validation through In Silico Cloning and Immune Simulation. Vaccines. 2021; 9(8):818. https://doi.org/10.3390/vaccines9080818
Chicago/Turabian StyleZheng, Bowen, Muhammad Suleman, Zonara Zafar, Syed Shujait Ali, Syed Nouman Nasir, Namra, Zahid Hussain, Muhammad Waseem, Abbas Khan, Fakhrul Hassan, and et al. 2021. "Towards an Ensemble Vaccine against the Pegivirus Using Computational Modelling Approaches and Its Validation through In Silico Cloning and Immune Simulation" Vaccines 9, no. 8: 818. https://doi.org/10.3390/vaccines9080818
APA StyleZheng, B., Suleman, M., Zafar, Z., Ali, S. S., Nasir, S. N., Namra, Hussain, Z., Waseem, M., Khan, A., Hassan, F., Wang, Y., & Wei, D. (2021). Towards an Ensemble Vaccine against the Pegivirus Using Computational Modelling Approaches and Its Validation through In Silico Cloning and Immune Simulation. Vaccines, 9(8), 818. https://doi.org/10.3390/vaccines9080818