
In Silico Identification of Cholesterol Binding Motifs in the Chemokine Receptor CCR3

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. CCR3 Homology Modeling and Structure Validation
2.2. Coarse-Grained Martini Simulations
2.3. Receptor–Lipid Analysis
2.4. Receptor Sequence Analysis
3. Results
3.1. Homology Modeling and Validation
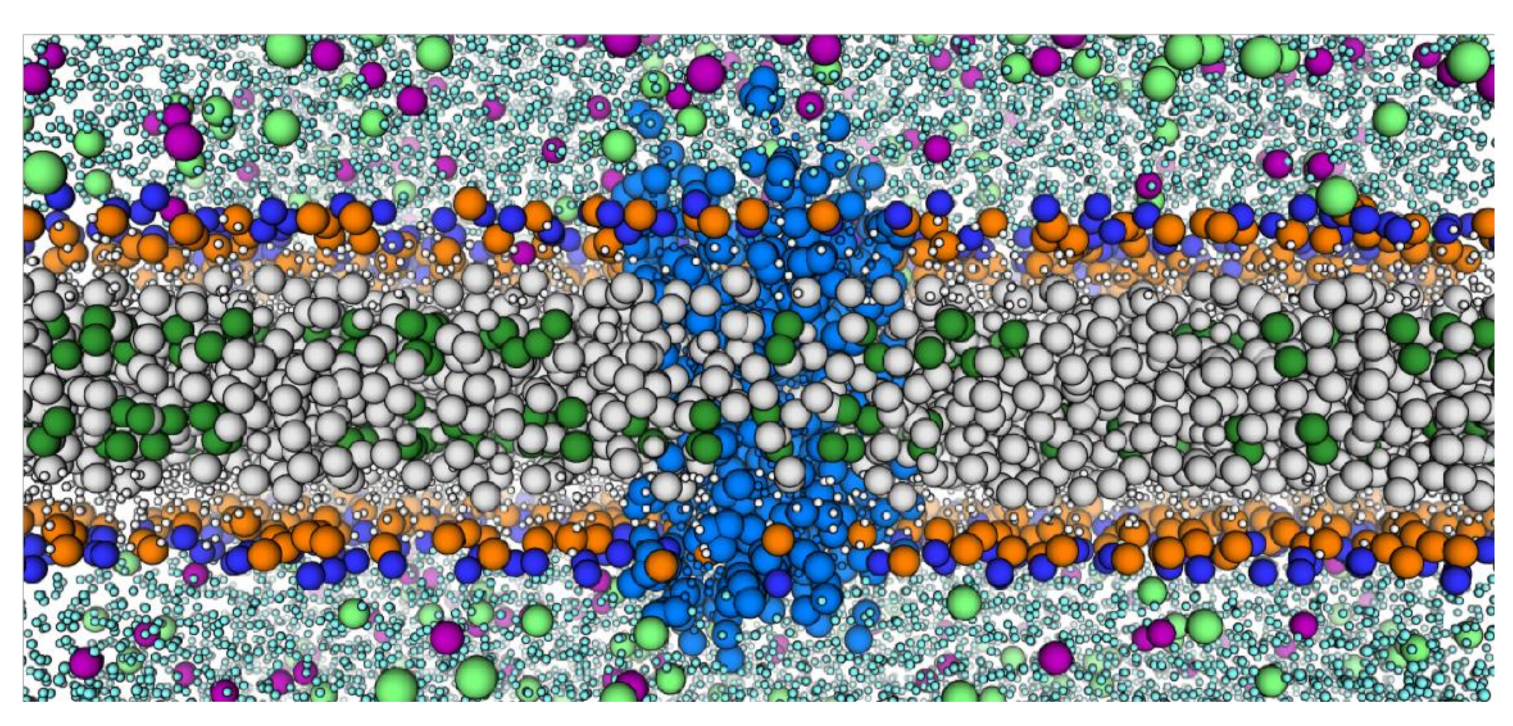
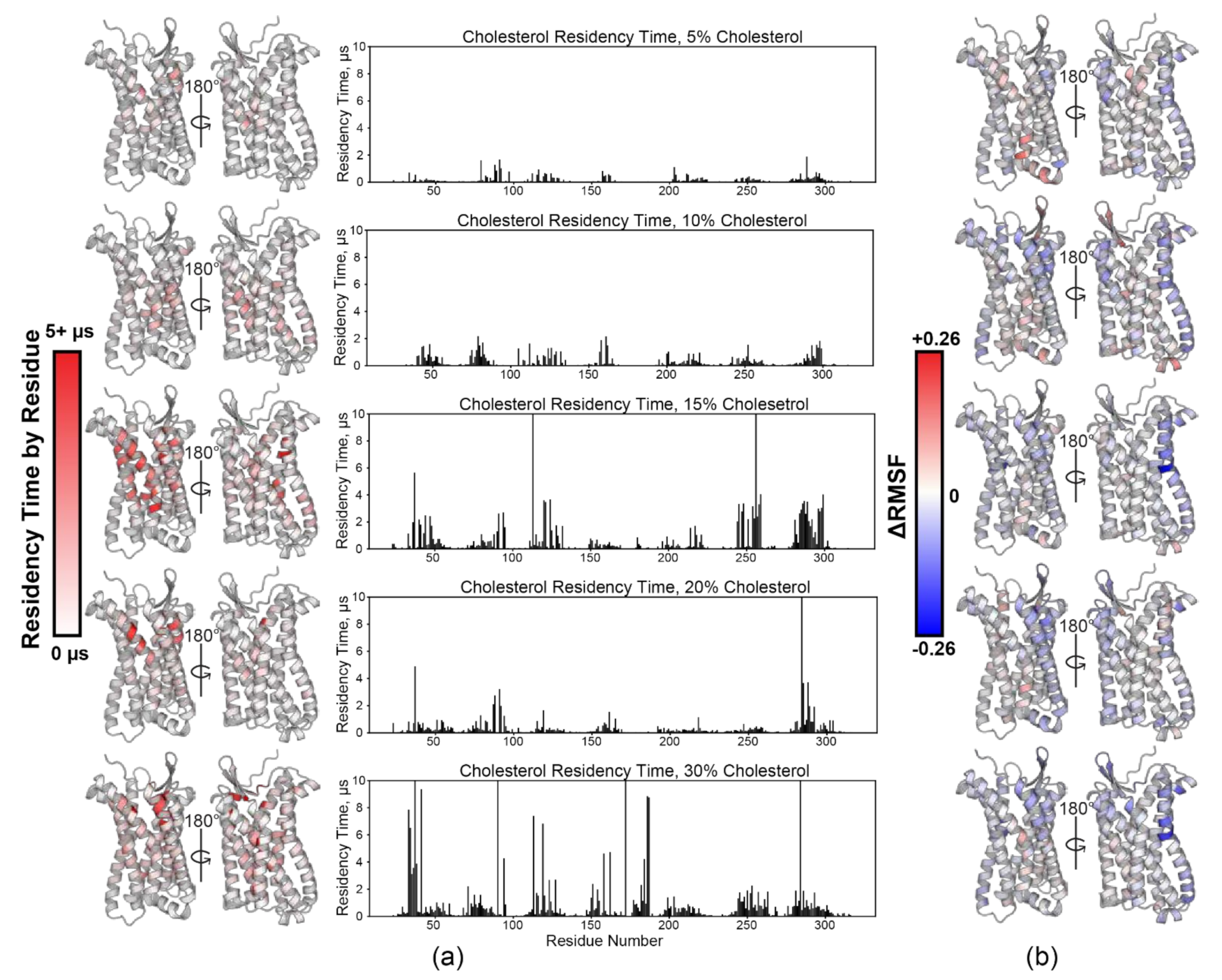
3.2. CCR3-Cholesterol Behavior in Simulation
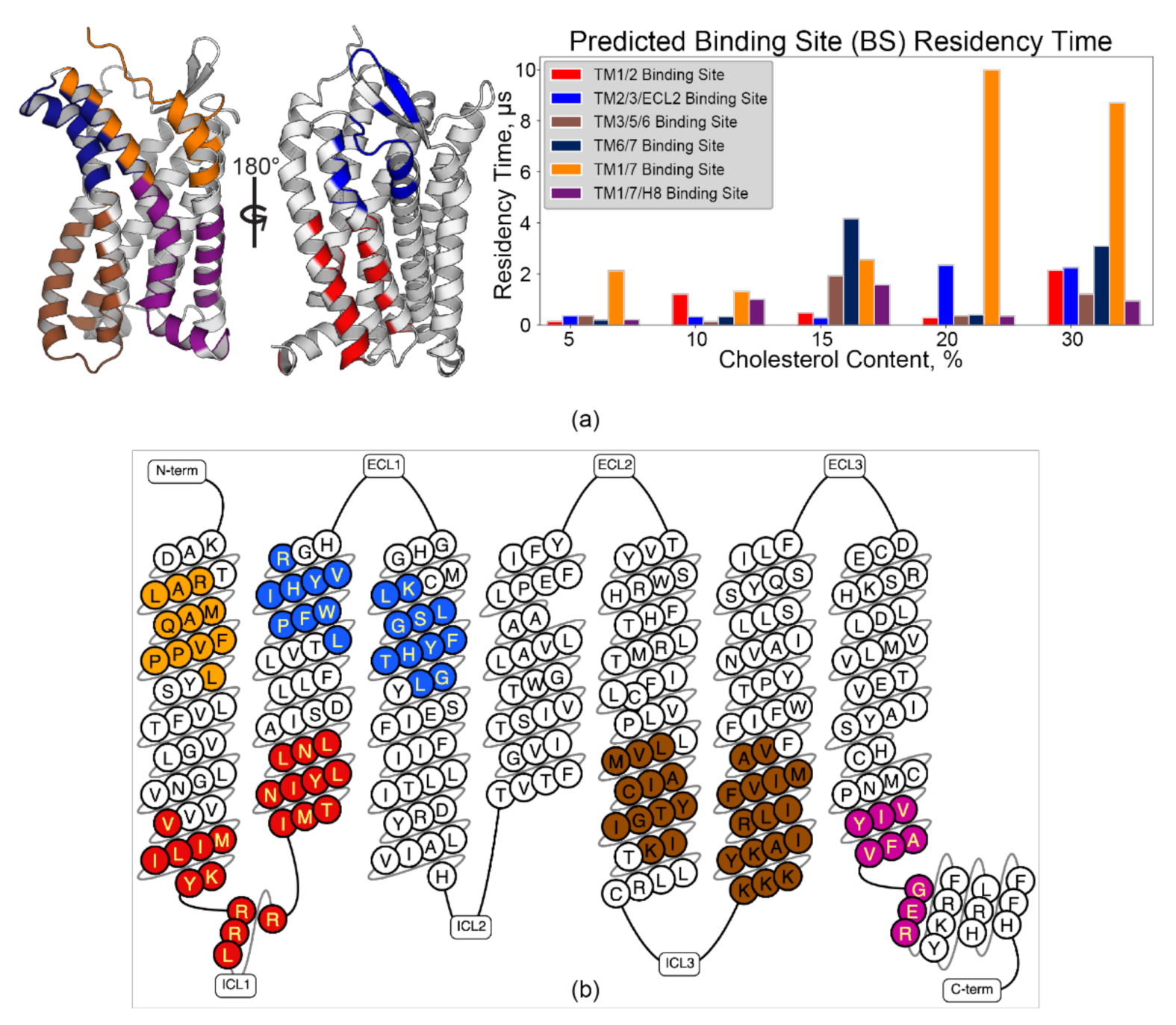
3.3. Pylipid Predicts Cholesterol Binding Sites in CCR3 Populated with CRAC and CARC Motifs
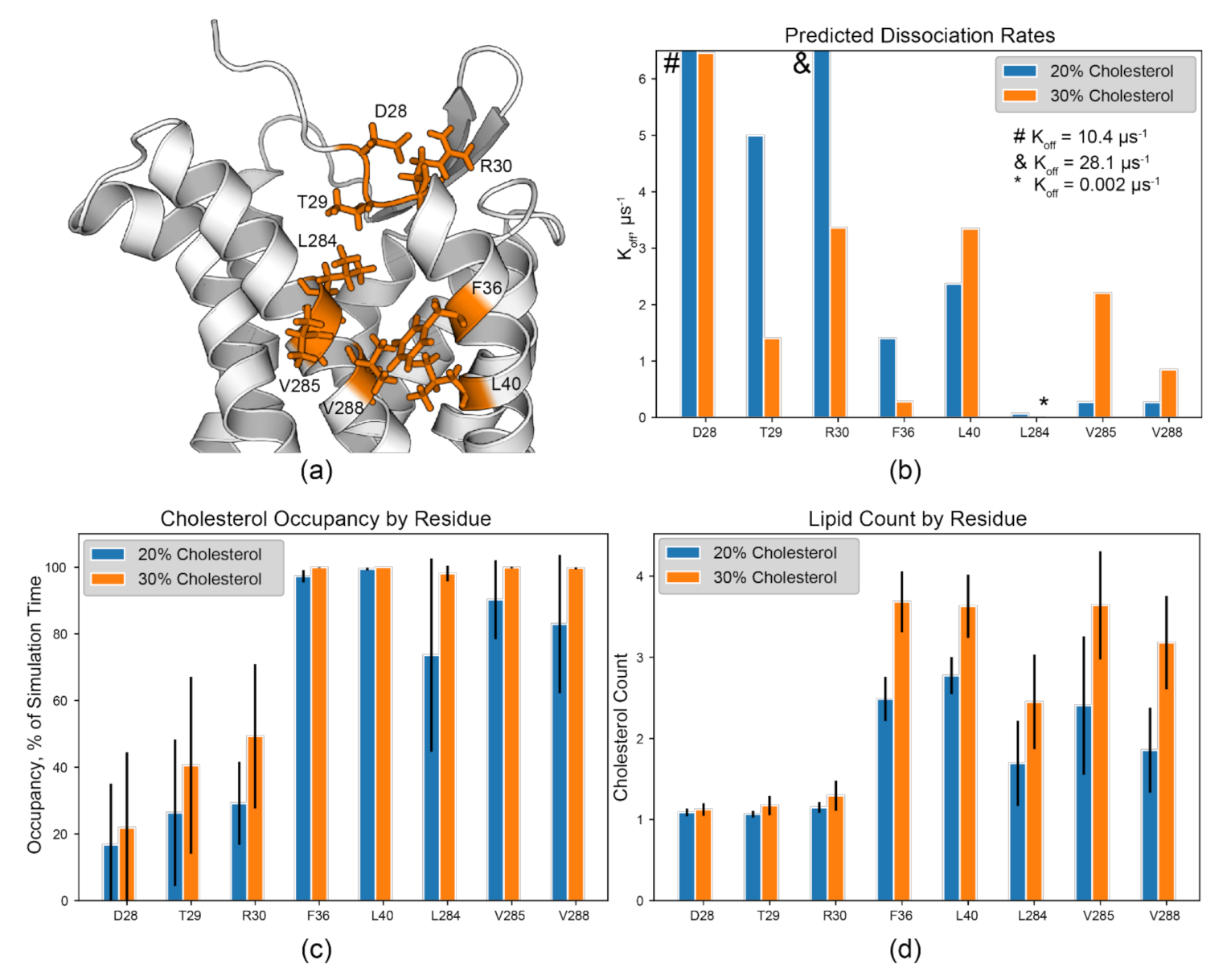
3.4. CARC-Like Motif in the TM1/7 Binding Site
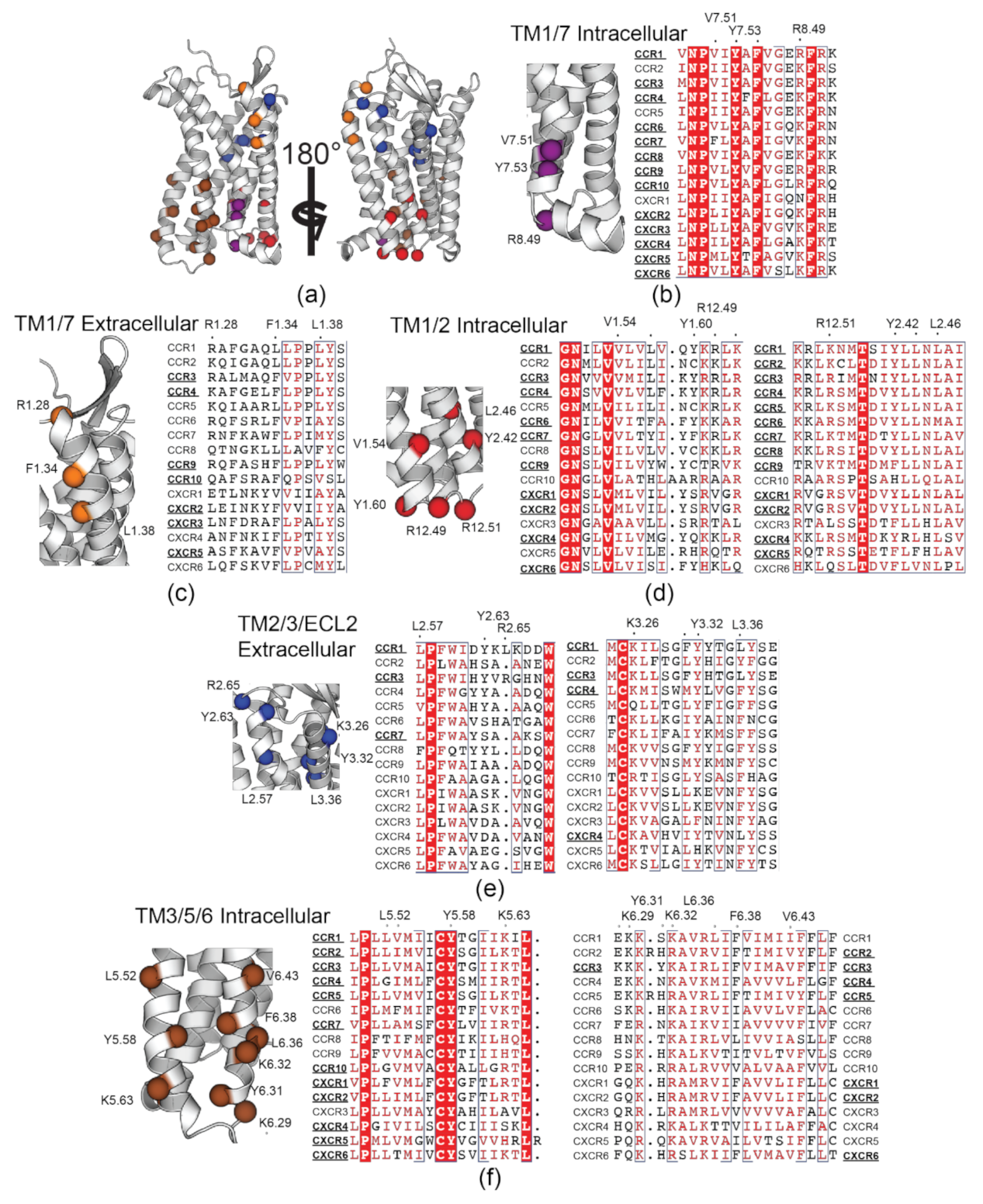
3.5. Rationaliztion of Identified CRAC/CARC Motif Conservation in Other Chemokine Receptors
4. Discussion
4.1. Cholesterol and Conformational Sampling
4.2. Curerent Experimental Evidence of CRAC/CARC Occupancy in Chemokine Receptorss
4.3. Cholesterol as Dynamic Glue Driving Multimerization
4.4. Pitfalls in Attempted Sorting of Cholesterol Binding Sites: Rejection of the Bear Thesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosenbaum, D.M.; Rasmussen, S.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nat. Cell Biol. 2009, 459, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Stone, M.J.; Hayward, J.; Huang, C.; Huma, Z.E.; Sanchez, J. Mechanisms of Regulation of the Chemokine-Receptor Network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef] [Green Version]
- Poeta, V.M.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Marzio, P.D.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nat. Cell Biol. 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.-P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; Van Antwerpen, P.; Kobilka, B.B.K.; Govaerts, C. Allosteric regulation of G protein–coupled receptor activity by phospholipids. Nat. Chem. Biol. 2015, 12, 35–39. [Google Scholar] [CrossRef]
- Yen, H.-Y.; Hoi, K.K.; Liko, I.; Hedger, G.; Horrell, M.R.; Song, W.; Wu, D.; Heine, P.; Warne, T.; Lee, Y.; et al. PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nat. Cell Biol. 2018, 559, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Calmet, P.; Cullin, C.; Cortès, S.; Vang, M.; Caudy, N.; Baccouch, R.; Dessolin, J.; Maamar, N.T.; LeComte, S.; Tillier, B.; et al. Cholesterol impacts chemokine CCR5 receptor ligand-binding activity. FEBS J. 2019, 287, 2367–2385. [Google Scholar] [CrossRef] [PubMed]
- Gahbauer, S.; Pluhackova, K.; Böckmann, R.A. Closely related, yet unique: Distinct homo- and heterodimerization patterns of G protein coupled chemokine receptors and their fine-tuning by cholesterol. PLoS Comput. Biol. 2018, 14, e1006062. [Google Scholar] [CrossRef]
- Zhukovsky, M.A.; Lee, P.-H.; Ott, A.; Helms, V. Putative cholesterol-binding sites in human immunodeficiency virus (HIV) coreceptors CXCR4 and CCR5. Proteins: Struct. Funct. Bioinform. 2012, 81, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Matti, C.; Laufer, J.M.; Jakobs, B.D.; Purvanov, V.; Allmen, E.U.-V.; Thelen, M. Modulation of Chemokine Receptor Function by Cholesterol: New Prospects for Pharmacological Intervention. Mol. Pharmacol. 2017, 91, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Pluhackova, K.; Gahbauer, S.; Kranz, F.; Wassenaar, T.A.; Böckmann, R.A. Dynamic Cholesterol-Conditioned Dimerization of the G Protein Coupled Chemokine Receptor Type 4. PLoS Comput. Biol. 2016, 12, e1005169. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yao, Z.-X.; Degenhardt, B.; Teper, G.; Papadopoulos, V. Cholesterol binding at the cholesterol recognition/ interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc. Natl. Acad. Sci. USA 2001, 98, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Epand, R.M.; Barrantes, F.J. Cholesterol-Recognition Motifs in Membrane Proteins. Adv. Exp. Med. Biol. 2019, 1135, 3–25. [Google Scholar] [CrossRef]
- Baier, C.J.; Fantini, J.; Barrantes, F.J. Disclosure of cholesterol recognition motifs in transmembrane domains of the human nicotinic acetylcholine receptor. Sci. Rep. 2011, 1, 69. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Schlegel, A.; Scherer, P.E.; Lisanti, M.P. Caveolins, a Family of Scaffolding Proteins for Organizing “Preassembled Signaling Complexes” at the Plasma Membrane. J. Biol. Chem. 1998, 273, 5419–5422. [Google Scholar] [CrossRef] [Green Version]
- Salzwedel, K.; West, J.T.; Hunter, E. A Conserved Tryptophan-Rich Motif in the Membrane-Proximal Region of the Human Immunodeficiency Virus Type 1 gp41 Ectodomain Is Important for Env-Mediated Fusion and Virus Infectivity. J. Virol. 1999, 73, 2469–2480. [Google Scholar] [CrossRef] [Green Version]
- Jafurulla, M.; Tiwari, S.; Chattopadhyay, A. Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein coupled receptors. Biochem. Biophys. Res. Commun. 2011, 404, 569–573. [Google Scholar] [CrossRef]
- Taghon, G.J.; Rowe, J.B.; Kapolka, N.J.; Isom, D.G. Predictable cholesterol binding sites in GPCRs lack consensus motifs. Structure 2021, 29, 499–506.e3. [Google Scholar] [CrossRef]
- Shihoya, W.; Nishizawa, T.; Yamashita, K.; Inoue, A.; Hirata, K.; Kadji, F.M.N.; Okuta, A.; Tani, K.; Aoki, J.; Fujiyoshi, Y.; et al. X-ray structures of endothelin ETB receptor bound to clinical antagonist bosentan and its analog. Nat. Struct. Mol. Biol. 2017, 24, 758–764. [Google Scholar] [CrossRef]
- Xing, C.; Zhuang, Y.; Xu, T.-H.; Feng, Z.; Zhou, X.E.; Chen, M.; Wang, L.; Meng, X.; Xue, Y.; Wang, J.; et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-Gi Signaling Complex. Cell 2020, 180, 645–654.e13. [Google Scholar] [CrossRef]
- Jakubík, J.; El-Fakahany, E. Allosteric Modulation of GPCRs of Class A by Cholesterol. Int. J. Mol. Sci. 2021, 22, 1953. [Google Scholar] [CrossRef]
- Sarkar, P.; Chattopadhyay, A. Cholesterol interaction motifs in G protein-coupled receptors: Slippery hot spots? Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1481. [Google Scholar] [CrossRef]
- Oswald, C.; Rappas, M.; Kean, J.; Doré, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; et al. Intracellular allosteric antagonism of the CCR9 receptor. Nat. Cell Biol. 2016, 540, 462–465. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Zhang, W.; et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Chien, E.Y.T.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Wu, L.; Yuan, S.; Wu, M.; Xu, Y.; Sun, Q.; Li, S.; Zhao, S.; Hua, T.; Liu, Z.-J. Structural basis of CXC chemokine receptor 2 activation and signalling. Nat. Cell Biol. 2020, 585, 1–9. [Google Scholar] [CrossRef]
- Ansell, T.B.; Song, W.; Sansom, M.S. The Glycosphingolipid GM3 Modulates Conformational Dynamics of the Glucagon Receptor. Biophys. J. 2020, 119, 300–313. [Google Scholar] [CrossRef]
- Duncan, A.; Corey, R.A.; Sansom, M.S.P. Defining how multiple lipid species interact with inward rectifier potassium (Kir2) channels. Proc. Natl. Acad. Sci. USA 2020, 117, 7803–7813. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.; Chattopadhyay, A. Identification of Cholesterol Binding Sites in the Serotonin1A Receptor. J. Phys. Chem. B 2012, 116, 12991–12996. [Google Scholar] [CrossRef]
- Ferraro, M.; Masetti, M.; Recanatini, M.; Cavalli, A.; Bottegoni, G. Mapping Cholesterol Interaction Sites on Serotonin Transporter through Coarse-Grained Molecular Dynamics. PLoS ONE 2016, 11, e0166196. [Google Scholar] [CrossRef]
- Horn, J.N.; Kao, T.-C.; Grossfield, A. Coarse-Grained Molecular Dynamics Provides Insight into the Interactions of Lipids and Cholesterol with Rhodopsin. Adv. Exp. Med. Biol. 2013, 796, 75–94. [Google Scholar] [CrossRef] [Green Version]
- Sejdiu, B.I.; Tieleman, D.P. Lipid-Protein Interactions Are a Unique Property and Defining Feature of G Protein-Coupled Receptors. Biophys. J. 2020, 118, 1887–1900. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; van Dijk, M.; de Vries, S.; Bonvin, A.M. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.C.T.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Cui, M.; Macalino, S.J.; Park, J.; Clavio, N.A.; Kang, S.; Choi, S. Exploring G Protein-Coupled Receptors (GPCRs) Ligand Space via Cheminformatics Approaches: Impact on Rational Drug Design. Front. Pharmacol. 2018, 9, 128. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Wang, M.; Li, J.; Liu, J.; Huang, F. Maltose binding protein facilitates functional production of engineered human chemokine receptor 3 in Escherichia coli. Process. Biochem. 2015, 50, 285–293. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; Fitzgerald, J.M.; Boulet, L.P.; Watson, R.M.; Hui, L.; Villineuve, H.; Scime, T.X.; Schlatman, A.R.; Obminski, C.; Kum, J.; et al. The effects of a CCR3 inhibitor, AXP1275, on allergen-induced airway responses in adults with mild-to-moderate atopic asthma. Clin. Exp. Allergy 2018, 48, 445–451. [Google Scholar] [CrossRef]
- Katschke, K.J., Jr.; Rottman, J.B.; Ruth, J.H.; Qin, S.; Wu, L.; LaRosa, G.; Ponath, P.; Park, C.C.; Pope, R.M.; Koch, A.E. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001, 44, 1022–1032. [Google Scholar] [CrossRef]
- Dunn, J.L.; Shoda, T.; Caldwell, J.M.; Wen, T.; Aceves, S.S.; Collins, M.H.; Dellon, E.S.; Falk, G.W.; Leung, J.; Martin, L.J.; et al. Esophageal type 2 cytokine expression heterogeneity in eosinophilic esophagitis in a multisite cohort. J. Allergy Clin. Immunol. 2020, 145, 1629–1640.e4. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Chen, Y.; Farzan, M.; Choe, H.; Ohagen, A.; Gartner, S.; Busciglio, J.; Yang, X.; Hofmann, W.; Newman, W.; et al. CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nat. Cell Biol. 1997, 385, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Liu, W.-R.; Tian, M.-X.; Jiang, X.-F.; Wang, H.; Zhou, P.-Y.; Ding, Z.-B.; Peng, Y.-F.; Dai, Z.; Qiu, S.-J.; et al. CCL24 contributes to HCC malignancy via RhoB- VEGFA-VEGFR2 angiogenesis pathway and indicates poor prognosis. Oncotarget 2016, 8, 5135–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Kido, A.; Akahane, M.; Kishi, S.; Tsukamoto, S.; Fujii, H.; Honoki, K.; Tanaka, Y. Mesenchymal stem cells up-regulate the invasive potential of prostate cancer cells via the eotaxin-3/CCR3 axis. Pathol. Res. Pr. 2018, 214, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Jöhrer, K.; Zelle-Rieser, C.; Perathoner, A.; Moser, P.; Hager, M.; Ramoner, R.; Gander, H.; Höltl, L.; Bartsch, G.; Greil, R.; et al. Up-Regulation of Functional Chemokine Receptor CCR3 in Human Renal Cell Carcinoma. Clin. Cancer Res. 2005, 11, 2459–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Periole, X.; Cavalli, M.; Marrink, S.; Ceruso, M.A. Combining an Elastic Network with a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef] [Green Version]
- De Jong, D.H.; Singh, G.; Bennett, D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2012, 9, 687–697. [Google Scholar] [CrossRef]
- Song, W.; Corey, R.A.; Duncan, A.L.; Ansell, T.B.; Stansfeld, P.J.; Sansom, M.S. Pylipid: A Python Toolkit for Analysis of Lipid-Protein Interactions from MD Simulations. Biophys. J. 2021, 120, 48a. [Google Scholar] [CrossRef]
- Van Aalst, E.J.; Wylie, B.J. Cholesterol is a dose-dependent positive allosteric modulator of CCR3 ligand affinity and G protein coupling. Front. Mol. Biosci. 2021. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.-R.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-Resolution Comparative Modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2017, 27, 293–315. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins: Struct. Funct. Bioinform. 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Wise, E.L.; Duchesnes, C.; da Fonseca, P.; Allen, R.A.; Williams, T.J.; Pease, J.E. Small Molecule Receptor Agonists and Antagonists of CCR3 Provide Insight into Mechanisms of Chemokine Receptor Activation. J. Biol. Chem. 2007, 282, 27935–27943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, Y.; Zhang, Y.; Dong, C.; Xu, B.; Sun, X. The small molecular CCR3 antagonist YM344031 attenuates neurodegenerative pathologies and improves learning and memory performance in a mouse model of Alzheimer’s disease. Brain Res. 2019, 1719, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Araújo, J.; Silva, L.; Felix, F.; Camargo, E.; Grespan, R. CCR3 antagonist impairs estradiol-induced eosinophil migration to the uterus in ovariectomized mice. Braz. J. Med. Biol. Res. 2020, 53, e8659. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Harris, R.; Olson, A.J.; Goodsell, D.S. Automated prediction of ligand-binding sites in proteins. Proteins Struct. Funct. Bioinform. 2007, 70, 1506–1517. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2015, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Ingólfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.; Im, W. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Bruininks, B.; Jefferies, D.; de Souza, P.C.T.; Lee, J.; Patel, D.S.; Marrink, S.J.; Qi, Y.; Khalid, S.; Im, W. CHARMM-GUI Martini Maker for modeling and simulation of complex bacterial membranes with lipopolysaccharides. J. Comput. Chem. 2017, 38, 2354–2363. [Google Scholar] [CrossRef] [Green Version]
- Knapp, B.; Ospina-Forero, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Hünenberger, P.H.; Van Gunsteren, W.F. Alternative schemes for the inclusion of a reaction-field correction into molecular dynamics simulations: Influence on the simulated energetic, structural, and dielectric properties of liquid water. J. Chem. Phys. 1998, 108, 6117–6134. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Maciejewski, M.W.; Schuyler, A.; Gryk, M.R.; Moraru, I.; Romero, P.R.; Ulrich, E.L.; Eghbalnia, H.R.; Livny, M.; Delaglio, F.; Hoch, J.C. NMRbox: A Resource for Biomolecular NMR Computation. Biophys. J. 2017, 112, 1529–1534. [Google Scholar] [CrossRef] [Green Version]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.; Mordalski, S.; Harpsøe, K.; Hauser, A.; Bojarski, A.; E Gloriam, D. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2017, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Han, G.W.; Abagyan, R.; Wu, B.; Stevens, R.; Cherezov, V.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 2017, 46, 1005–1017.e5. [Google Scholar] [CrossRef] [Green Version]
- Wassenaar, T.A.; Ingólfsson, H.I.; Boeckmann, R.A.; Tieleman, D.P.; Marrink, S. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 2144–2155. [Google Scholar] [CrossRef]
- Marrink, S.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.N.; I Ingolfsson, H.; Marrink, S. Parameters for Martini sterols and hopanoids based on a virtual-site description. J. Chem. Phys. 2015, 143, 243152. [Google Scholar] [CrossRef]
- Jones, A.J.Y.; Gabriel, F.; Tandale, A.; Nietlispach, D. Structure and Dynamics of GPCRs in Lipid Membranes: Physical Principles and Experimental Approaches. Molecules 2020, 25, 4729. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Zhang, H.; Mao, C.; Zhou, X.E.; Cheng, X.; Simon, I.A.; Shen, D.-D.; Yen, H.-Y.; Robinson, C.V.; et al. Structural insights into the lipid and ligand regulation of serotonin receptors. Nat. Cell Biol. 2021, 592, 469–473. [Google Scholar] [CrossRef]
- Prioleau, C.; Visiers, I.; Ebersole, B.J.; Weinstein, H.; Sealfon, S. Conserved Helix 7 Tyrosine Acts as a Multistate Conformational Switch in the 5HT2C Receptor. J. Biol. Chem. 2002, 277, 36577–36584. [Google Scholar] [CrossRef] [Green Version]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 2018, 172, 68–80.e12. [Google Scholar] [CrossRef] [Green Version]
- Miao, Y.; Caliman, A.D.; McCammon, J.A. Allosteric Effects of Sodium Ion Binding on Activation of the M3 Muscarinic G-Protein-Coupled Receptor. Biophys. J. 2015, 108, 1796–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.L.; Eddy, M.T.; Gao, Z.-G.; Han, G.W.; Lian, T.; Deary, A.; Patel, N.; Jacobson, K.; Katritch, V.; Stevens, R.C. Structural Connection between Activation Microswitch and Allosteric Sodium Site in GPCR Signaling. Structure 2018, 26, 259–269.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.A.; Weinstein, H. [19] Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar] [CrossRef]
- Schönegge, A.-M.; Gallion, J.; Picard, L.-P.; Wilkins, A.D.; Le Gouill, C.; Audet, M.; Stallaert, W.; Lohse, M.; Kimmel, M.; Lichtarge, O.; et al. Evolutionary action and structural basis of the allosteric switch controlling β2AR functional selectivity. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Picard, L.-P.; Schönegge, A.-M.; Bouvier, M. Structural Insight into G Protein-Coupled Receptor Signaling Efficacy and Bias between Gs and β-Arrestin. ACS Pharmacol. Transl. Sci. 2019, 2, 148–154. [Google Scholar] [CrossRef]
- Rasmussen, S.; DeVree, B.; Zou, Y.; Kruse, A.C.; Chung, K.Y.; Kobilka, T.S.; Thian, F.S.; Chae, P.S.; Pardon, E.; Calinski, D.; et al. Crystal structure of the β2 adrenergic receptor–Gs protein complex. Nat. Cell Biol. 2011, 477, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Wacker, D.; Wang, C.; Katritch, V.; Han, G.W.; Huang, X.-P.; Vardy, E.; McCorvy, J.D.; Jiang, Y.; Chu, M.; Siu, F.Y.; et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Wasilko, D.J.; Johnson, Z.L.; Ammirati, M.; Che, Y.; Griffor, M.C.; Han, S.; Wu, H. Structural basis for chemokine receptor CCR6 activation by the endogenous protein ligand CCL20. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Dalton, J.A.R.; Lans, I.; Giraldo, J. Quantifying conformational changes in GPCRs: Glimpse of a common functional mechanism. BMC Bioinform. 2015, 16, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, K.K.; Shalev-Benami, M.; Robertson, M.J.; Hu, H.; Banister, S.; Hollingsworth, S.A.; Latorraca, N.R.; Kato, H.; Hilger, D.; Maeda, S.; et al. Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 2019, 176, 448–458.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gusach, A.; Luginina, A.; Marin, E.; Brouillette, R.L.; Besserer-Offroy, É.; Longpré, J.M.; Ishchenko, A.; Popov, P.; Patel, N.; Fujimoto, T.; et al. Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Chen, T.; Xiong, M.; Zong, X.; Ge, Y.; Zhang, H.; Wang, M.; Han, G.W.; Yi, C.; Ma, L.; Ye, R.D.; et al. Structural basis of ligand binding modes at the human formyl peptide receptor 2. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Ge, B.; Lao, J.; Wang, Z.; Yang, B.; Wang, X.; He, H.; Li, J.; Huang, F. Regulation of the Oligomeric Status of CCR3 with Binding Ligands Revealed by Single-Molecule Fluorescence Imaging. Biochemistry 2017, 57, 852–860. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Momboisse, F.; Boncompain, G.; Koensgen, F.; Zhou, Z.; Cordeiro, N.; Arenzana-Seisdedos, F.; Perez, F.; Lagane, B.; Kellenberger, E.; et al. CCR5 adopts three homodimeric conformations that control cell surface delivery. Sci. Signal. 2018, 11, eaal2869. [Google Scholar] [CrossRef] [Green Version]
- Salanga, C.L.; O’Hayre, M.; Handel, T. Modulation of chemokine receptor activity through dimerization and crosstalk. Cell. Mol. Life Sci. 2008, 66, 1370–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddese, B.; Deniaud, M.; Garnier, A.; Tiss, A.; Guissouma, H.; Abdi, H.; Henrion, D.; Chabbert, M. Evolution of chemokine receptors is driven by mutations in the sodium binding site. PLoS Comput. Biol. 2018, 14, e1006209. [Google Scholar] [CrossRef]
- Pelé, J.; Abdi, H.; Moreau, M.; Thybert, D.; Chabbert, M. Multidimensional Scaling Reveals the Main Evolutionary Pathways of Class A G-Protein-Coupled Receptors. PLoS ONE 2011, 6, e19094. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Aalst, E.; Koneri, J.; Wylie, B.J. In Silico Identification of Cholesterol Binding Motifs in the Chemokine Receptor CCR3. Membranes 2021, 11, 570. https://doi.org/10.3390/membranes11080570
van Aalst E, Koneri J, Wylie BJ. In Silico Identification of Cholesterol Binding Motifs in the Chemokine Receptor CCR3. Membranes. 2021; 11(8):570. https://doi.org/10.3390/membranes11080570
Chicago/Turabian Stylevan Aalst, Evan, Jotham Koneri, and Benjamin J. Wylie. 2021. "In Silico Identification of Cholesterol Binding Motifs in the Chemokine Receptor CCR3" Membranes 11, no. 8: 570. https://doi.org/10.3390/membranes11080570
APA Stylevan Aalst, E., Koneri, J., & Wylie, B. J. (2021). In Silico Identification of Cholesterol Binding Motifs in the Chemokine Receptor CCR3. Membranes, 11(8), 570. https://doi.org/10.3390/membranes11080570