

Mechanism of Action and Structure–Activity Relationships of Tetracyclic Small Molecules Acting as Universal Positive Allosteric Modulators of the Cholecystokinin Receptor
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
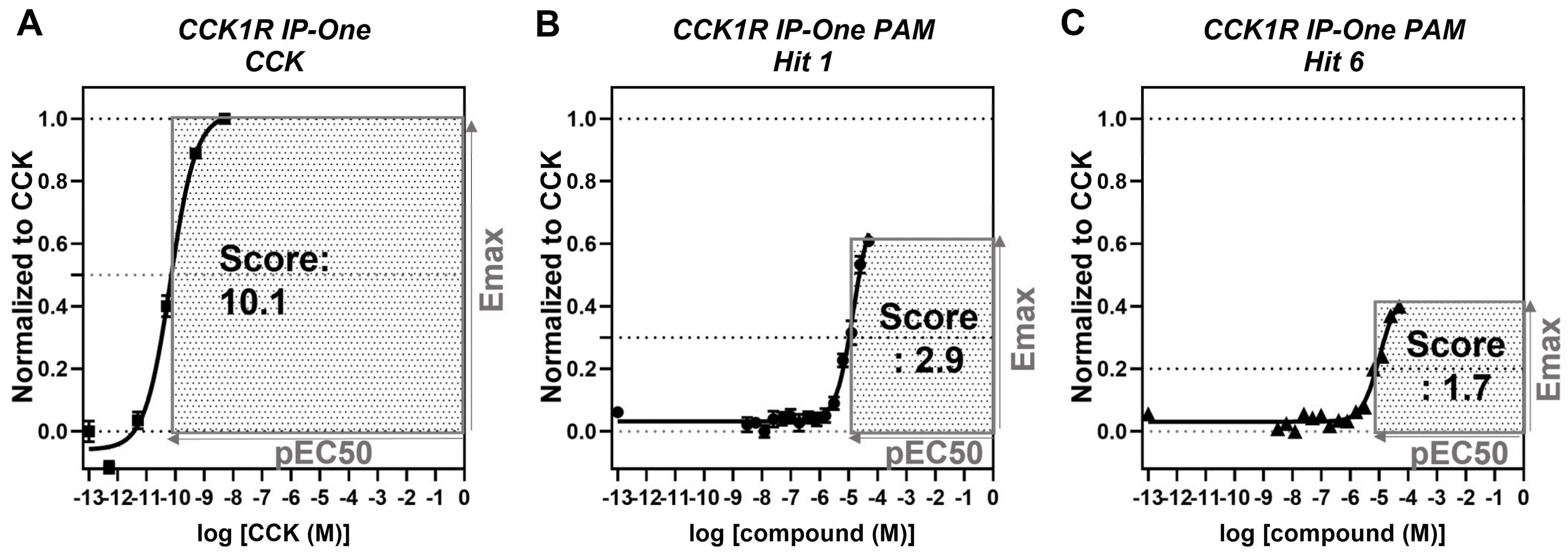
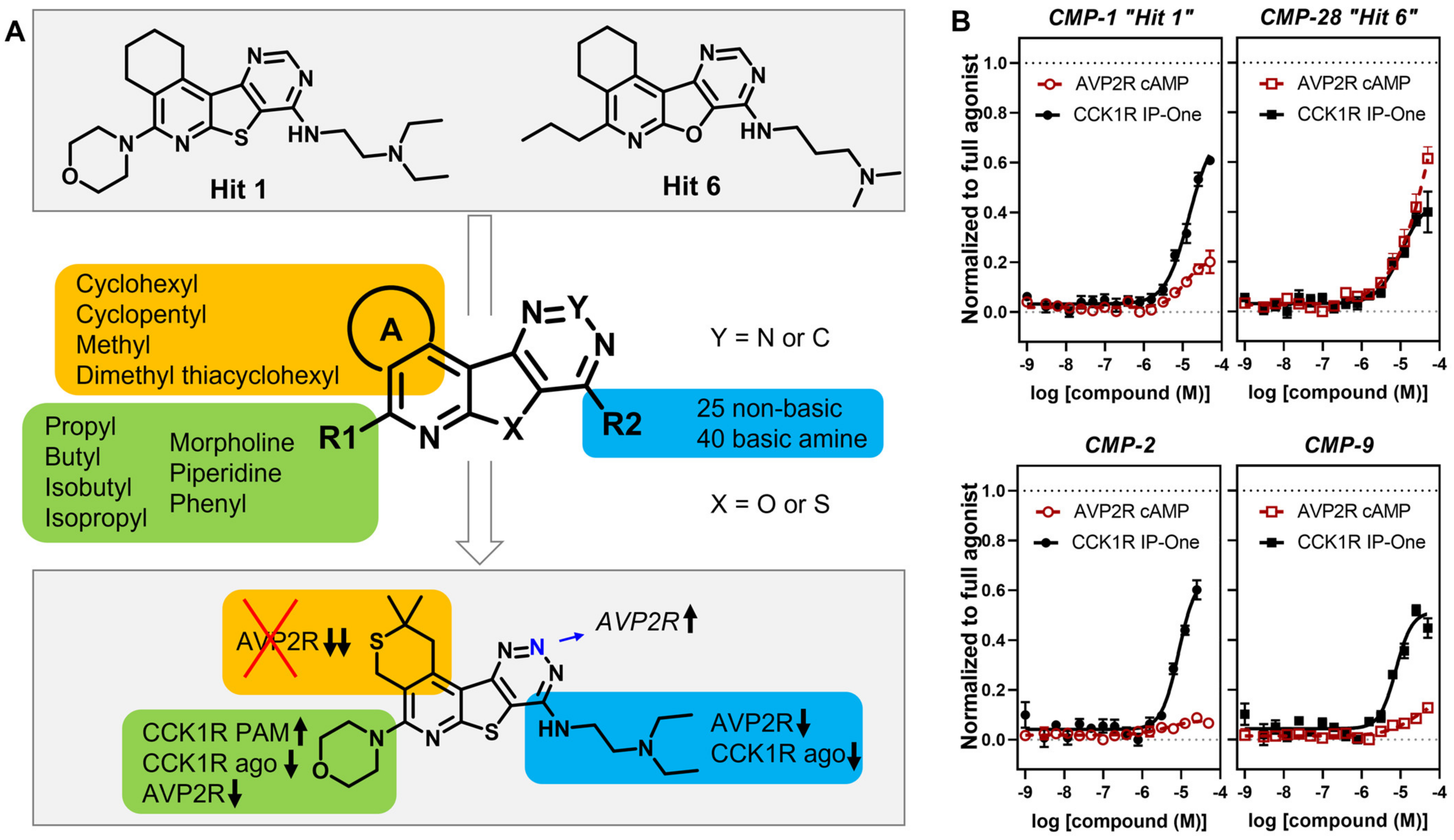
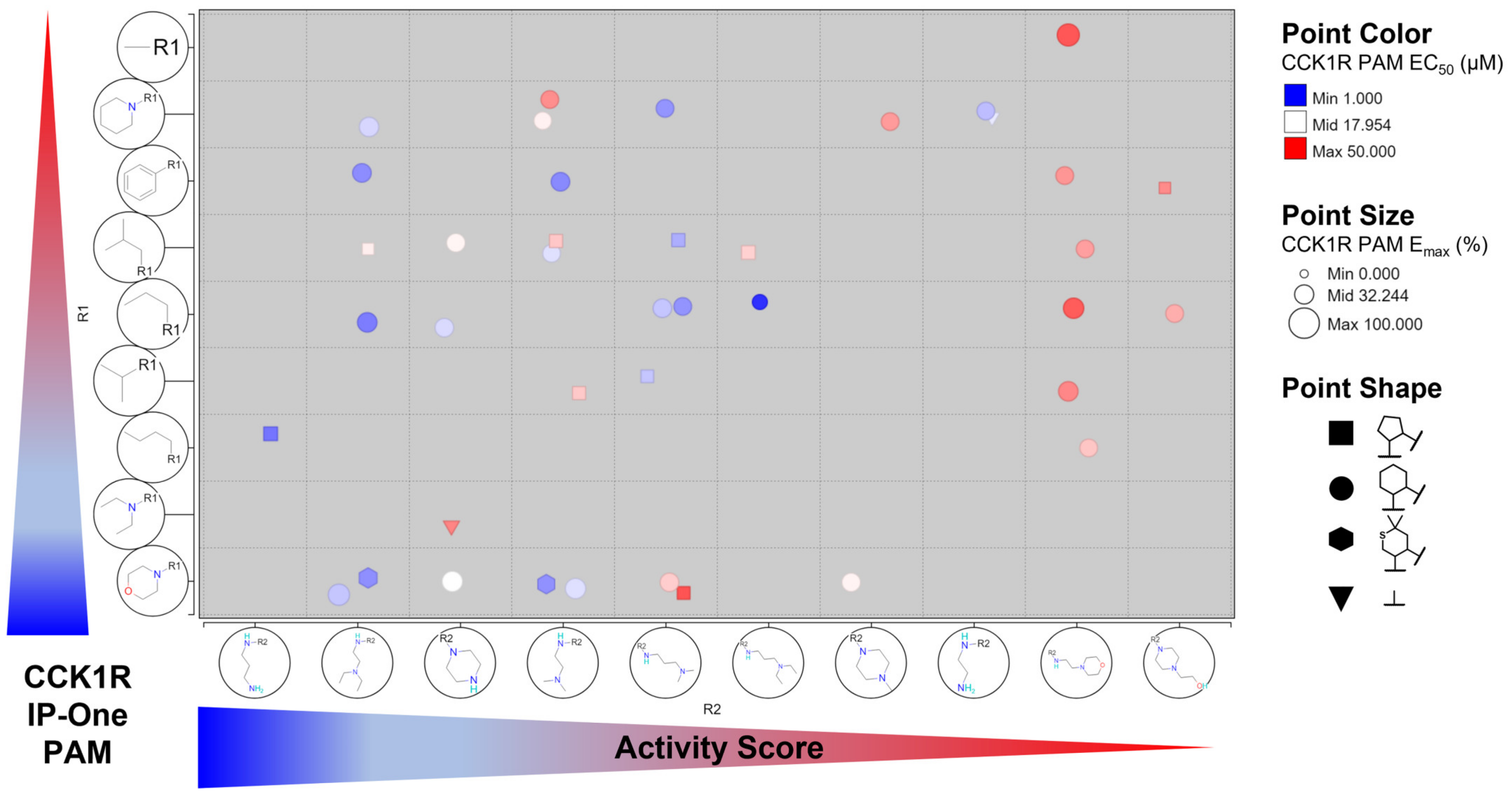
3. Results
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, G.P.; Gibbs, J. The satiety effect of cholecystokinin. Recent progress and current problems. Ann. N. Y. Acad. Sci. 1985, 448, 417–423. [Google Scholar] [CrossRef]
- Desai, A.J.; Dong, M.; Harikumar, K.G.; Miller, L.J. Cholecystokinin-induced satiety, a key gut servomechanism that is affected by the membrane microenvironment of this receptor. Int. J. Obes. Suppl. 2016, 6, S22–S27. [Google Scholar] [CrossRef]
- Miller, L.J.; Desai, A.J. Metabolic actions of the type 1 cholecystokinin receptor: Its potential as a therapeutic target. Trends Endocrinol. Metab. 2016, 27, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.; Greenway, F.; Leiter, L.; Li, Z.; Jacobson, P.; Murphy, K.; Hill, J.; Kler, L.; Aftring, R. Stimulation of cholecystokinin-A receptors with GI181771X does not cause weight loss in overweight or obese patients. Clin. Pharmacol. Ther. 2008, 83, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.; Harikumar, K.; Wootten, D.; Sexton, P. Roles of Cholecystokinin in the Nutritional Continuum. Physiology and Potential Therapeutics. Front. Endocrinol. 2021, 12, 684656. [Google Scholar] [CrossRef] [PubMed]
- Dengler, D.; Sun, Q.; Harikumar, K.; Miller, L.; Sergienko, E. Screening for positive allosteric modulators of cholecystokinin type 1 receptor potentially useful for management of obesity. SLAS Discov. 2022, 27, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.; Pinon, D.; Miller, L. Use of N,O-bis-Fmoc-D-Tyr-ONSu for introduction of an oxidative iodination site into cholecystokinin family peptides. Int. J. Pept. Protein Res. 1988, 31, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Harikumar, K.; Pinon, D.; Wessels, W.; Prendergast, F.; Miller, L. Environment and mobility of a series of fluorescent reporters at the amino terminus of structurally related peptide agonists and antagonists bound to the cholecystokinin receptor. J. Biol. Chem. 2002, 277, 18552–18560. [Google Scholar] [CrossRef] [PubMed]
- Hadac, E.; Ghanekar, D.; Holicky, E.; Pinon, D.; Dougherty, R.; Miller, L. Relationship between native and recombinant cholecystokinin receptors: Role of differential glycosylation. Pancreas 1996, 13, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Potter, R.; Harikumar, K.; Wu, S.; Miller, L. Differential sensitivity of types 1 and 2 cholecystokinin receptors to membrane cholesterol. J. Lipid Res. 2012, 53, 137–148. [Google Scholar] [CrossRef]
- Desai, A.; Harikumar, K.; Miller, L. A type 1 cholecystokinin receptor mutant that mimics the dysfunction observed for wild type receptor in a high cholesterol environment. J. Biol. Chem. 2014, 289, 18314–18326. [Google Scholar] [CrossRef]
- Harikumar, K.; Coudrat, T.; Desai, A.; Dong, M.; Dengler, D.; Furness, S.; Christopoulos, A.; Wootten, D.; Sergienko, E.; Sexton, P.; et al. Discovery of a Positive Allosteric Modulator of Cholecystokinin Action at CCK1R in Normal and Elevated Cholesterol. Front. Endocrinol. 2021, 12, 89957. [Google Scholar] [CrossRef]
- Kim, G.; Lin, J.; Blomain, E.; Waldman, S. Antiobesity pharmacotherapy: New drugs and emerging targets. Clin. Pharmacol. Ther. 2014, 95, 53–66. [Google Scholar] [CrossRef]
- Rueda-Clausen, C.; Padwal, R.; Sharma, A. New pharmacological approaches for obesity management. Nat. Rev. Endocrinol. 2013, 9, 467–478. [Google Scholar] [CrossRef]
- Pathak, V.; Flatt, P.; Irwin, N. Cholecystokinin (CCK) and related adjunct peptide therapies for the treatment of obesity and type 2 diabetes. Peptides 2018, 100, 229–235. [Google Scholar] [CrossRef]
- Piper, N.; Whitfield, E.; Stewart, G.; Xu, X.; Furness, S. Targeting appetite and satiety in diabetes and obesity, via G protein-coupled receptors. Biochem. Pharmacol. 2022, 202, 115115. [Google Scholar] [CrossRef]
- Muller, T.; Bluher, M.; Tschop, M.; DiMarchi, R. Anti-obesity drug discovery: Advances and challenges. Nat. Rev. Drug. Discov. 2022, 21, 201–223. [Google Scholar] [CrossRef]
- Chandra, R.; Liddle, R. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes Obes. 2007, 14, 63–67. [Google Scholar] [CrossRef]
- Sherrill, R.; Berman, J.; Birkemo, L.; Croom, D.; Dezube, M.; Ervin, G.; Grizzle, M.; James, M.; Johnson, M.; Queen, K.; et al. 1,4-Benzodiazepine peripheral cholecystokinin (CCK-A) receptor agonists. Bioorg. Med. Chem. Lett. 2001, 11, 1145–1148. [Google Scholar] [CrossRef]
- Berger, R.; Zhu, C.; Hansen, A.; Harper, B.; Chen, Z.; Holt, T.; Hubert, J.; Lee, S.; Pan, J.; Qian, S.; et al. 2-Substituted piperazine-derived imidazole carboxamides as potent and selective CCK1R agonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2008, 18, 4833–4837. [Google Scholar] [CrossRef]
- Zhu, C.; Hansen, A.; Bateman, T.; Chen, Z.; Holt, T.; Hubert, J.; Karanam, B.; Lee, S.; Pan, J.; Qian, S.; et al. Discovery of imidazole carboxamides as potent and selective CCK1R agonists. Bioorg. Med. Chem. Lett. 2008, 18, 4393–4396. [Google Scholar] [CrossRef]
- Sensfuss, U.; Kruse, T.; Skyggebjerg, R.; Uldam, H.; Vestergaard, B.; Huus, K.; Vinther, T.; Reinau, M.; Scheele, S.; Clausen, T. Structure-activity relationships and characterization of highly selective, long-acting, peptide-based cholecystokinin 1 receptor agonists. J. Med. Chem. 2019, 62, 1407–1419. [Google Scholar] [CrossRef]
- Dawra, R.; Saluja, A.; Lerch, M.; Saluja, M.; Logsdon, C.; Steer, M. Stimulation of pancreatic growth by cholecystokinin is mediated by high affinity receptors on rat pancreatic acinar cells. Biochem. Biophys. Res. Commun. 1993, 193, 814–820. [Google Scholar] [CrossRef]
- Hoshi, H.; Logsdon, C. Both low- and high-affinity CCK receptor states mediate trophic effects on rat pancreatic acinar cells. Am. J. Physiol. 1993, 265, G1177–G1181. [Google Scholar] [CrossRef]
- Gitto, S.; Nakkina, S.; Beardsley, J.; Parikh, J.; Altomare, D. Induction of pancreatitis in mice with susceptibility to pancreatic cancer. Methods Cell Biol. 2022, 168, 139–159. [Google Scholar]
- Christopoulos, A. Advances in g protein-coupled receptor allostery: From function to structure. Mol. Pharmacol. 2014, 86, 463–478. [Google Scholar] [CrossRef]
- Paragh, G.; Kovacs, E.; Seres, I.; Keresztes, T.; Balogh, Z.; Szabo, J.; Teichmann, F.; Foris, G. Altered signal pathway in granulocytes from patients with hypercholesterolemia. J. Lipid Res. 1999, 40, 1728–1733. [Google Scholar] [CrossRef]
- Seres, I.; Foris, G.; Varga, Z.; Kosztaczky, B.; Kassai, A.; Balogh, Z.; Fulop, P.; Paragh, G. The association between angiotensin II-induced free radical generation and membrane fluidity in neutrophils of patients with metabolic syndrome. J. Membr. Biol. 2006, 214, 91–98. [Google Scholar] [CrossRef]
- Kenakin, T. Theoretical aspects of GPCR-ligand complex pharmacology. Chem. Rev. 2017, 117, 4–20. [Google Scholar] [CrossRef]
- Mobbs, J.; Belousoff, M.; Harikumar, K.; Piper, S.; Xu, X.; Furness, S.; Venugopal, H.; Christopoulos, A.; Danev, R.; Wootten, D.; et al. Structures of the human cholecystokinin 1 (CCK1) receptor bound to Gs and Gq mimetic proteins provide insight into mechanisms of G protein selectivity. PLoS Biol. 2021, 19, e3001295. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
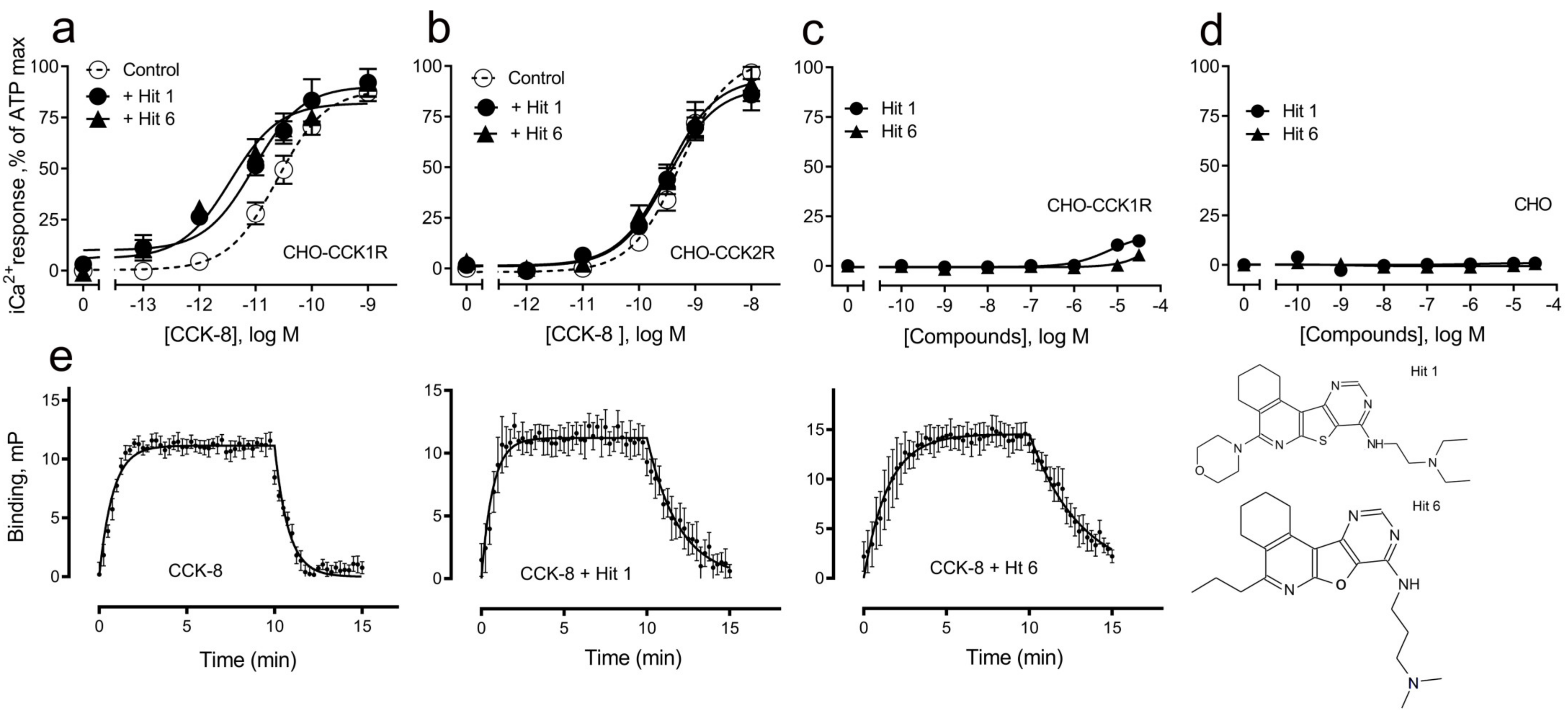
| Receptor-Ligands | pEC50 | n, p Values |
|---|---|---|
| CCK1R | ||
| CCK-8 | 10.6 ± 0.1 | 5 |
| CCK-8 + “hit 1” | 11.1 ± 0.1 * | 5, 0.048 |
| CCK-8 + “hit 6” | 11.5 ± 0.2 ** | 5, 0.002 |
| CCK1R | ||
| CCK-33 | 9.6 ± 0.2 | 5 |
| CCK-33 + “hit 1” | 10.7 ± 0.2 * | 5, 0.016 |
| CCK-33 + “hit 6” | 10.7 ± 0.2 ** | 5, 0.008 |
| CCK2R | ||
| CCK-8 | 9.3 ± 0.1 | 6 |
| CCK-8 + “hit 1” | 9.5 ± 0.1 | 6, 0.20 |
| CCK-8 + “hit 6” | 9.5 ± 0.1 | 6, 0.19 |
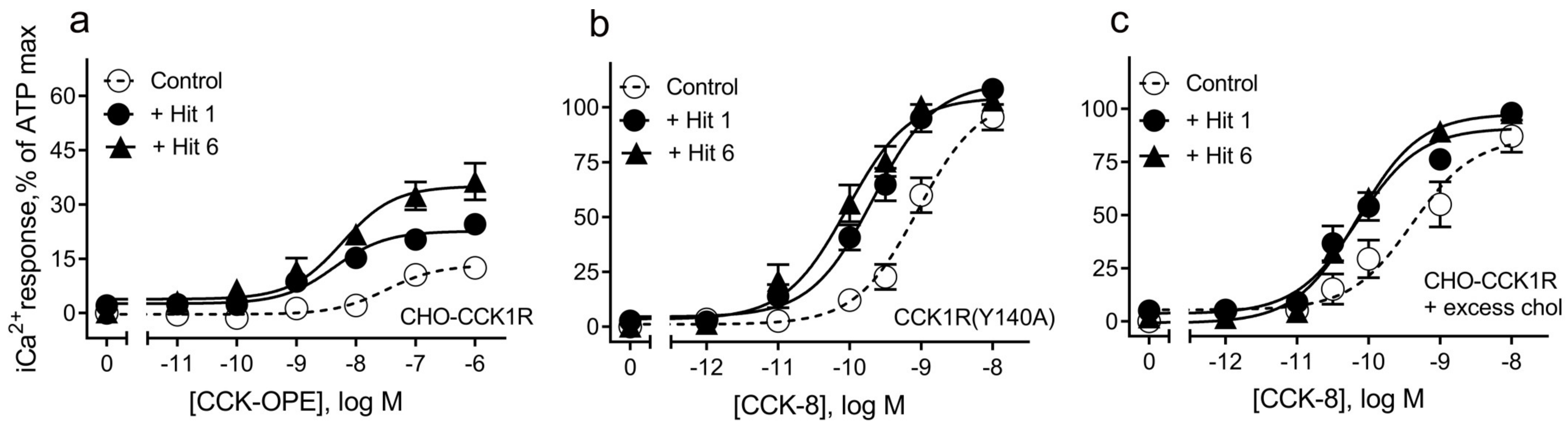
| CCK1R(Y140A) | ||
| CCK-8 | 9.0 ± 0.1 | 5 |
| CCK-8 + “hit 1” | 9.7 ± 0.1 *** | 5, 0.0008 |
| CCK-8 + “hit 6” | 10 ± 0.1 *** | 5, <0.0001 |
| CCK1R | ||
| CCK-OPE | 7.3 ± 0.2 | 5 |
| CCK-OPE + “hit 1” | 8.0 ± 0.2 | 5, 0.08 |
| CCK-OPE + “hit 6” | 8.1 ± 0.2 * | 6, 0.04 |
| CCK1R + excess cholesterol | ||
| CCK-8 | 9.4 ± 0.2 | 5 |
| CCK-8 + “hit 1” | 10.2 ± 0.2 ** | 5, 0.005 |
| CCK-8 + “hit 6” | 10.2 ± 0.2 ** | 6, 0.005 |
| CCK-8 | n | CCK-8 + “Hit 1” | n, p Values | CCK-8 + “Hit 6” | n, p Values | |
|---|---|---|---|---|---|---|
| Kon rate, × 108 M−1 min−1 | 0.7 ± 0.1 | 5 | 2.6 ± 0.6 * | 3, 0.04 | 0.7 ± 0.4 | 5, 0.31 |
| Koff rate, min−1 | 1.1 ± 0.1 | 5 | 0.4 ± 0.1 * | 3, 0.04 | 0.5 ± 0.1 ** | 5, 0.01 |
| pKi | 7.8 ± 0.1 | 5 | 8.8 ± 0.1 * | 3, 0.04 | 8.1 ± 0.2 | 5, 0.06 |
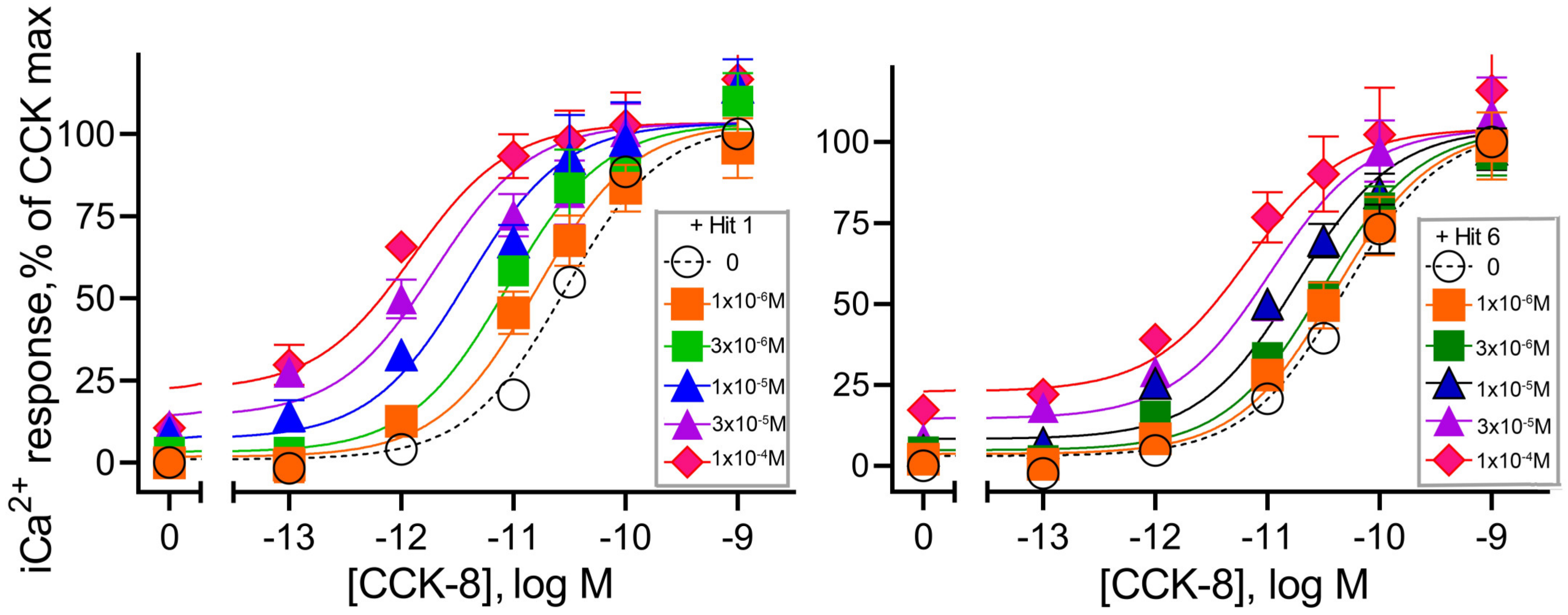
| CCK-8 + “Hit 1” | CCK-8 + “Hit 6” | |
|---|---|---|
| pKb | 4.5 ± 0.4 | 4.6 ± 0.2 |
| Tau Kb | 0.5 ± 0.1 | 0.4 ± 0.1 |
| Logαβ | 1.5 ± 0.3 | 1.0 ± 0.2 |
| n | 5 | 5 |
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Structure | CCK1R PAM (IP-One) | CCK1R Ago (IP-One) | AVP2R PAM (cAMP) | ||||||||||
| R1 | R2 | A | X | Y | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | |
| CMP-40 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 2.9 ± 0.1 | 96 ± 9 | 5.33 |
| CMP-41 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-65 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 10.0 ± 1.7 | 96 ± 5 | 4.78 |
| CMP-42 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-44 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-45 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 37.9 ± 7.0 | 33 ± 13 | 1.46 |
| CMP-64 |  |  | cyclohexyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-46 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-47 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-48 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-49 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-50 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-51 |  |  | cyclohexyl | S | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-52 |  |  | cyclohexyl | S | N | 0 | 0 | 0 | 0 | 0 | 0 | N.D. | N.D. | N.D. |
| CMP-59 |  |  | cyclohexyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 22.3 ± 7.2 | 20 ± 0 | 0.93 |
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Structure | CCK1R PAM (IP-One) | CCK1R Ago (IP-One) | AVP2R PAM (cAMP) | ||||||||||
| R1 | R2 | A | X | Y | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | |
| CMP-56 |  |  | cyclopentyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-57 |  |  | cyclohexyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-58 |  |  | cyclohexyl | O | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-60 |  |  | cyclohexyl | O | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-38 |  |  | cyclohexyl | S | C | 7.6 ± 3.3 | 20 ± 7.6 | 1.01 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-62 |  |  | cyclopentyl | O | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-39 |  |  | cyclohexyl | O | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-63 |  |  | cyclopentyl | O | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-55 |  |  | cyclohexyl | O | C | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| CMP-53 |  |  | cyclohexyl | O | C | 0 | 0 | 0 | 0 | 0 | 0 | 15.3 ± 9.8 | 61 ± 12 | 2.95 |
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Structure | CCK1R PAM (IP-One) | CCK1R ago (IP-One) | AVP2R PAM (cAMP) | ||||||||||
| R1 | R2 | A | X | Y | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | |
| CMP-5 |  |  | cyclopentyl | S | C | 7.5 ± 2.9 | 53 ± 6 | 2.66 | N.D. | N.D. | N.D. | 8.1 ± 8.5 | 32 ± 11 | 1.61 |
| CMP-7 |  |  | cyclohexyl | S | C | 8.0 ± 2.1 | 48 ± 11 | 2.39 | 0 | 0 | 0 | 6.5 ± 4.2 | 31 ± 2 | 1.61 |
| CMP-8 |  |  | cyclohexyl | S | C | 37.8 ± 3.9 | 53 ± 1 | 2.34 | 0 | 0 | 0 | 15.7 ± 8.4 | 48 ± 12 | 2.31 |
| CMP-12 |  |  | cyclohexyl | O | C | 14.1 ± 4.3 | 46 ± 7 | 2.23 | N.D. | N.D. | N.D. | 11.4 ± 3.6 | 66 ± 3 | 3.28 |
| CMP-18 |  |  | cyclohexyl | S | C | 12.7 ± 5.5 | 43 ± 21 | 1.96 | 0 | 0 | 0 | 11.8 ± 14.0 | 40 ± 17 | 1.99 |
| CMP-28 (“hit 6”) |  |  | cyclohexyl | O | C | 9.5 ± 3.5 | 35 ± 13 | 1.66 | N.D. | N.D. | N.D. | 15.0 ± 13.6 | 73 ± 16 | 3.53 |
| CMP-30 |  |  | cyclohexyl | O | C | 27.1 ± 10.7 | 36 ± 16 | 1.55 | 0 | 0 | 0 | 5.9 ± 3.8 | 29 ± 0 | 1.52 |
| CMP-32 |  |  | cyclohexyl | S | C | 23.9 ± 25.8 | 41 ± 34 | 1.52 | 0 | 0 | 0 | 9.3 ± 11.1 | 33 ± 26 | 1.67 |
| CMP-36 |  |  | cyclohexyl | O | C | 3.3 ± 5.4 | 25 ± 10 | 1.32 | 0 | 0 | 0 | 6.1 ± 2.8 | 27 ± 20 | 1.41 |
| CMP-61 |  |  | cyclohexyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 0 | 0 | 0 |
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Structure | CCK1R PAM (IP-One) | CCK1R ago (IP-One) | AVP2R PAM (cAMP) | ||||||||||
| R1 | R2 | A | X | Y | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | |
| CMP-10 |  |  | cyclopentyl | O | C | 22.5 ± 9.0 | 51 ± 16 | 2.28 | N.D. | N.D. | N.D. | 21.7 ± 11.3 | 46 ± 9 | 2.15 |
| CMP-13 |  |  | cyclopentyl | O | C | 11.2 ± 4.2 | 52 ± 36 | 2.23 | 0 | 0 | 0 | 12.6 ± 7.9 | 28 ± 14 | 1.35 |
| CMP-15 |  |  | cyclohexyl | O | C | 17.8 ± 6.2 | 43 ± 3 | 2.04 | 0 | 0 | 0 | 7.3 ± 3.3 | 58 ± 15 | 2.97 |
| CMP-20 |  |  | cyclopentyl | O | C | 23.6 ± 1.0 | 42 ± 12 | 1.90 | 0 | 0 | 0 | 21.2 ± 19.7 | 25 ± 1 | 1.17 |
| CMP-21 |  |  | cyclopentyl | O | C | 23.8 ± 10.5 | 45 ± 23 | 1.90 | 0 | 0 | 0 | 22.8 ± 3.8 | 37 ± 4 | 1.74 |
| CMP-24 |  |  | cyclopentyl | O | C | 12.7 ± 15.5 | 48 ± 45 | 1.81 | 0 | 0 | 0 | 19.6 ± 5.1 | 34 ± 23 | 1.62 |
| CMP-25 |  |  | cyclohexyl | O | C | 14.5 ± 5.3 | 38 ± 11 | 1.79 | N.D. | N.D. | N.D. | 8.6 ± 5.4 | 74 ± 12 | 3.75 |
| CMP-33 |  |  | cyclopentyl | O | C | 18.8 ± 7.7 | 35 ± 19 | 1.51 | 0 | 0 | 0 | 21.0 ± 6.5 | 34 ± 1 | 1.60 |
| CMP-34 |  |  | cyclohexyl | S | C | 29.0 ± 13.1 | 34 ± 13 | 1.50 | 0 | 0 | 0 | 7.8 ± 4.0 | 42 ± 17 | 2.13 |
| CMP-54 |  |  | cyclohexyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | 26.2 ± 2.4 | 43 ± 7 | 1.96 |
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Structure | CCK1R PAM (IP-One) | CCK1R ago (IP-One) | AVP2R PAM (cAMP) | ||||||||||
| R1 | R2 | A | X | Y | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | |
| CMP-6 |  |  | cyclohexyl | S | C | 13.8 ± 3.2 | 57 ± 27 | 2.58 | 0 | 0 | 0 | 9.9 ± 0.5 | 33 ± 1 | 1.67 |
| CMP-11 |  |  | cyclohexyl | S | C | 9.1 ± 4.8 | 45 ± 6 | 2.27 | 27.1 ± 15.3 | 21 ± 2 | 0.96 | 3.2 ± 1.1 | 44 ± 8 | 2.44 |
| CMP-14 |  |  | cyclohexyl | S | C | 8.9 ± 1.7 | 44 ± 10 | 2.17 | 29.7 ± 8.0 | 17 ± 5 | 0.72 | 4.7 ± 4.1 | 56 ± 33 | 2.97 |
| CMP-22 |  |  | cyclohexyl | S | C | 12.2 ± 4.6 | 39 ± 11 | 1.87 | 26.9 ± 1.1 | 60 ± 4.5 | 2.76 | 2.7 ± 0.7 | 52 ± 4 | 2.92 |
| CMP-23 |  |  | cyclohexyl | S | N | 9.6 ± 3.6 | 38 ± 13 | 1.86 | 30.0 ± 4.9 | 41 ± 1 | 1.85 | 3.1 ± 1.3 | 53 ± 3 | 2.93 |
| CMP-26 |  |  | cyclohexyl | S | C | 30.2 ± 11.2 | 41 ± 15 | 1.76 | N.D. | N.D. | N.D. | 3.7 ± 0.5 | 16 ± 4 | 0.87 |
| CMP-27 |  |  | cyclohexyl | S | N | 18.4 ± 8.1 | 38 ± 14 | 1.70 | 0 | 0 | 0 | 9.1 ± 1.0 | 70 ± 4 | 3.51 |
| CMP-29 |  |  | 4-methyl | S | C | 15.1 ± 1.8 | 33 ± 7 | 1.59 | 52.9 ± 39 | 39 ± 12 | 1.67 | 19.2 ± 0.8 | 35 ± 0 | 1.66 |
| CMP-31 |  |  | cyclohexyl | S | C | 29.6 ± 1.8 | 35 ± 14 | 1.54 | 0 | 0 | 0 | 5.4 ± 1.7 | 24 ± 3 | 1.26 |
| CMP-35 |  |  | cyclohexyl | S | C | 31.3 ± 14.9 | 36 ± 20 | 1.49 | 0 | 0 | 0 | 7.9 ± 2.5 | 31 ± 0 | 1.59 |
| CMP-37 |  |  | cyclopentyl | S | C | 31.7 ± 1.2 | 28 ± 9 | 1.26 | 0 | 0 | 0 | 0 | 0 | 0 |
 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Structure | CCK1R PAM (IP-One) | CCK1R Ago (IP-One) | AVP2R PAM (cAMP) | ||||||||||
| R1 | R2 | A | X | Y | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | EC50 (µM) | Emax (%) | Score | |
| CMP-1 (“hit 1”) |  |  | cyclohexyl | S | C | 12.8 ± 4.6 | 63 ± 18 | 2.94 | N.D. | N.D. | N.D. | 12.0 ± 7.1 | 20.7 ± 9.8 | 1.02 |
| CMP-2 |  |  | dimethyl thiacyclohe-xyl | S | C | 9.2 ± 2.0 | 57 ± 12 | 2.82 | 31.3 ± 3.1 | 31 ± 15 | 1.31 | N.D. | N.D. | N.D. |
| CMP-3 |  |  | cyclohexyl | S | C | 16.6 ± 15.9 | 60 ± 23 | 2.77 | N.D. | N.D. | N.D. | 14.8 ± 10.3 | 34.0 ± 5.3 | 1.64 |
| CMP-4 |  |  | cyclohexyl | S | C | 14.4 ± 1.5 | 62 ± 33 | 2.71 | 0 | 0 | 0 | 12.8 ± 14.0 | 24.5 ± 7.1 | 1.20 |
| CMP-9 |  |  | dimethyl thiacyclohe-xyl | S | C | 9.3 ± 1.1 | 46 ± 3 | 2.31 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. |
| CMP-16 |  |  | methyl | S | C | 32.6 ± 12.5 | 45 ± 7 | 2.02 | 0 | 0 | 0 | 7.6 ± 8.2 | 17 ± 4 | 0.87 |
| CMP-17 |  |  | cyclohexyl | S | N | 23.0 ± 17.9 | 50 ± 32 | 1.99 | N.D. | N.D. | N.D. | 22.4 ± 9.2 | 26 ± 1 | 1.20 |
| CMP-19 |  |  | cyclohexyl | S | C | 18.2 ± 8.6 | 51 ± 43 | 1.94 | 0 | 0 | 0 | N.D. | N.D. | N.D. |
| CMP-43 |  |  | cyclopentyl | S | C | N.D. | N.D. | N.D. | 0 | 0 | 0 | N.D. | N.D. | N.D. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dengler, D.G.; Harikumar, K.G.; Yen, A.; Sergienko, E.A.; Miller, L.J. Mechanism of Action and Structure–Activity Relationships of Tetracyclic Small Molecules Acting as Universal Positive Allosteric Modulators of the Cholecystokinin Receptor. Membranes 2023, 13, 150. https://doi.org/10.3390/membranes13020150
Dengler DG, Harikumar KG, Yen A, Sergienko EA, Miller LJ. Mechanism of Action and Structure–Activity Relationships of Tetracyclic Small Molecules Acting as Universal Positive Allosteric Modulators of the Cholecystokinin Receptor. Membranes. 2023; 13(2):150. https://doi.org/10.3390/membranes13020150
Chicago/Turabian StyleDengler, Daniela G., Kaleeckal G. Harikumar, Alice Yen, Eduard A. Sergienko, and Laurence J. Miller. 2023. "Mechanism of Action and Structure–Activity Relationships of Tetracyclic Small Molecules Acting as Universal Positive Allosteric Modulators of the Cholecystokinin Receptor" Membranes 13, no. 2: 150. https://doi.org/10.3390/membranes13020150
APA StyleDengler, D. G., Harikumar, K. G., Yen, A., Sergienko, E. A., & Miller, L. J. (2023). Mechanism of Action and Structure–Activity Relationships of Tetracyclic Small Molecules Acting as Universal Positive Allosteric Modulators of the Cholecystokinin Receptor. Membranes, 13(2), 150. https://doi.org/10.3390/membranes13020150