
Restricted Surface Diffusion of Cytochromes on Bioenergetic Membranes with Anionic Lipids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
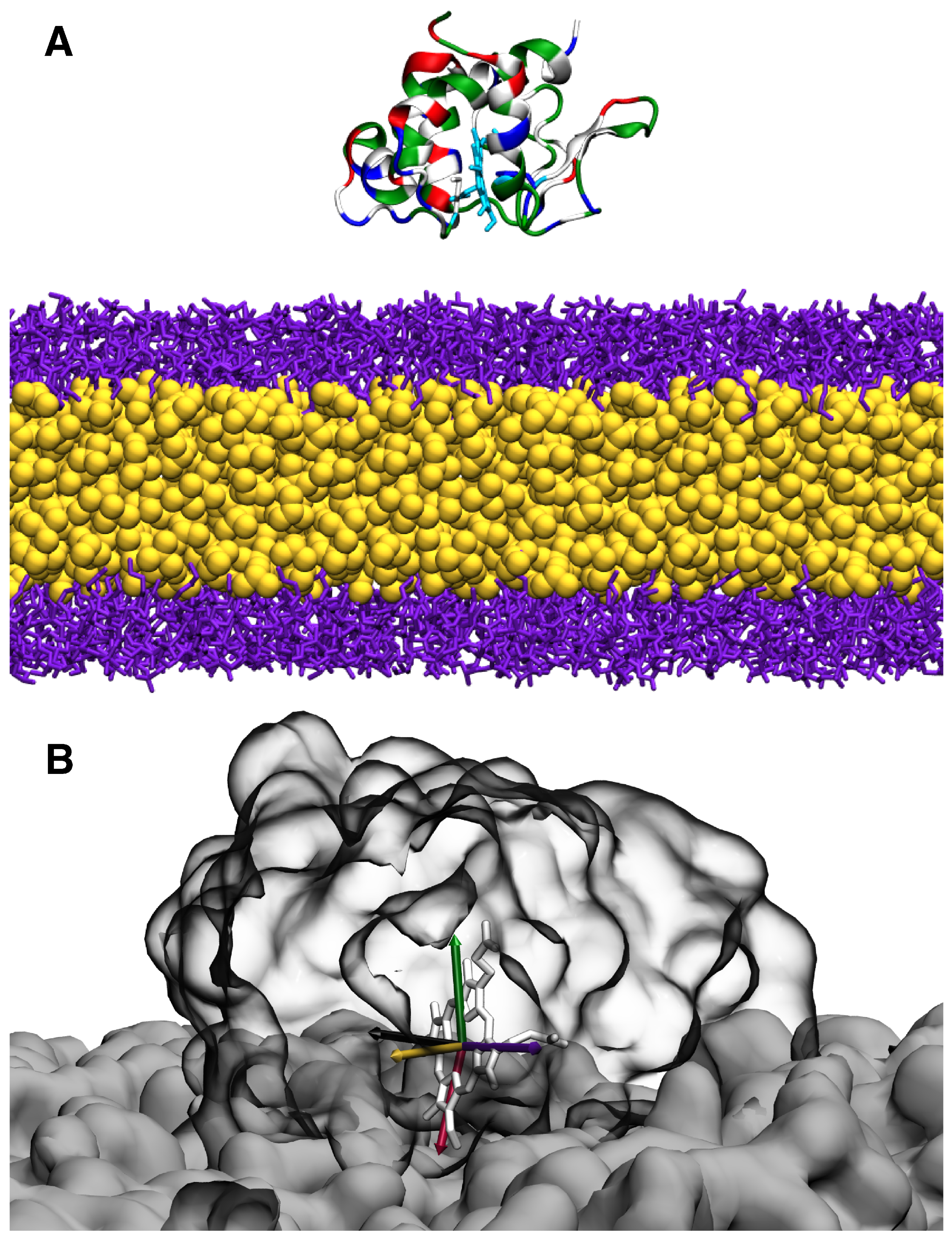
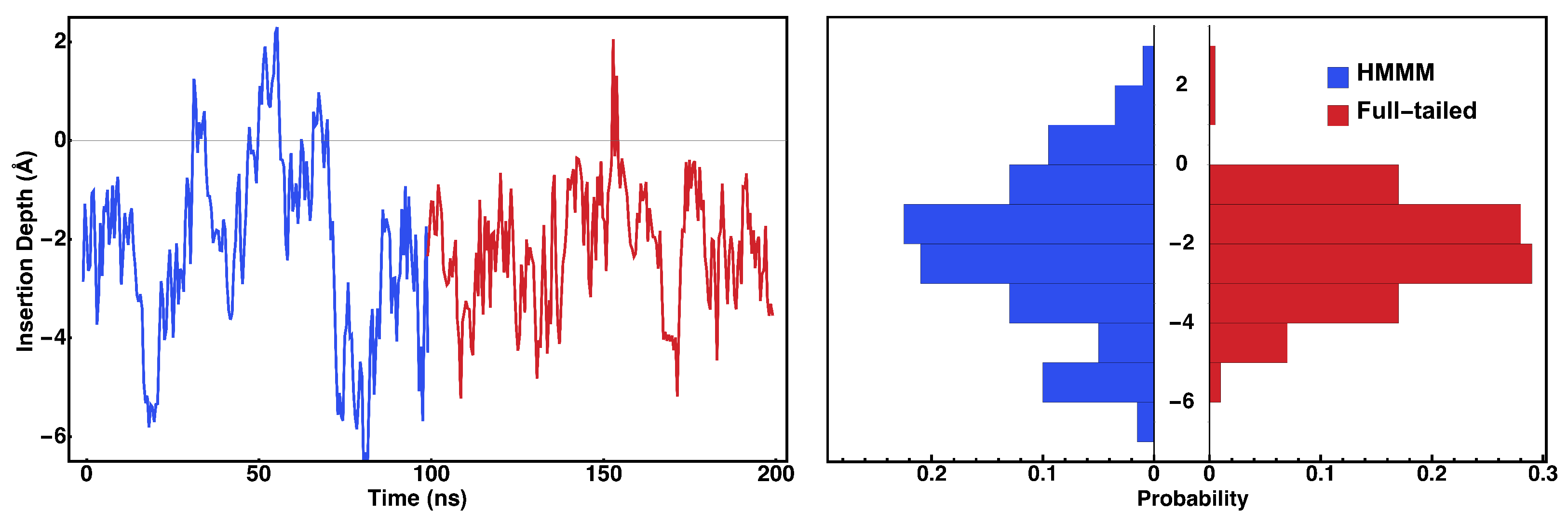
2.1. HMMM Simulation System
2.2. Characterization of the Membrane-Bound State of cyt c2
- : Angle made by the membrane normal (z-axis) and normal axis of the heme ring.
- : Angle made by the membrane and heme co-normal with the vector in the plane of the heme ring.
- : Angle made by the membrane and heme co-normal with the x-axis.
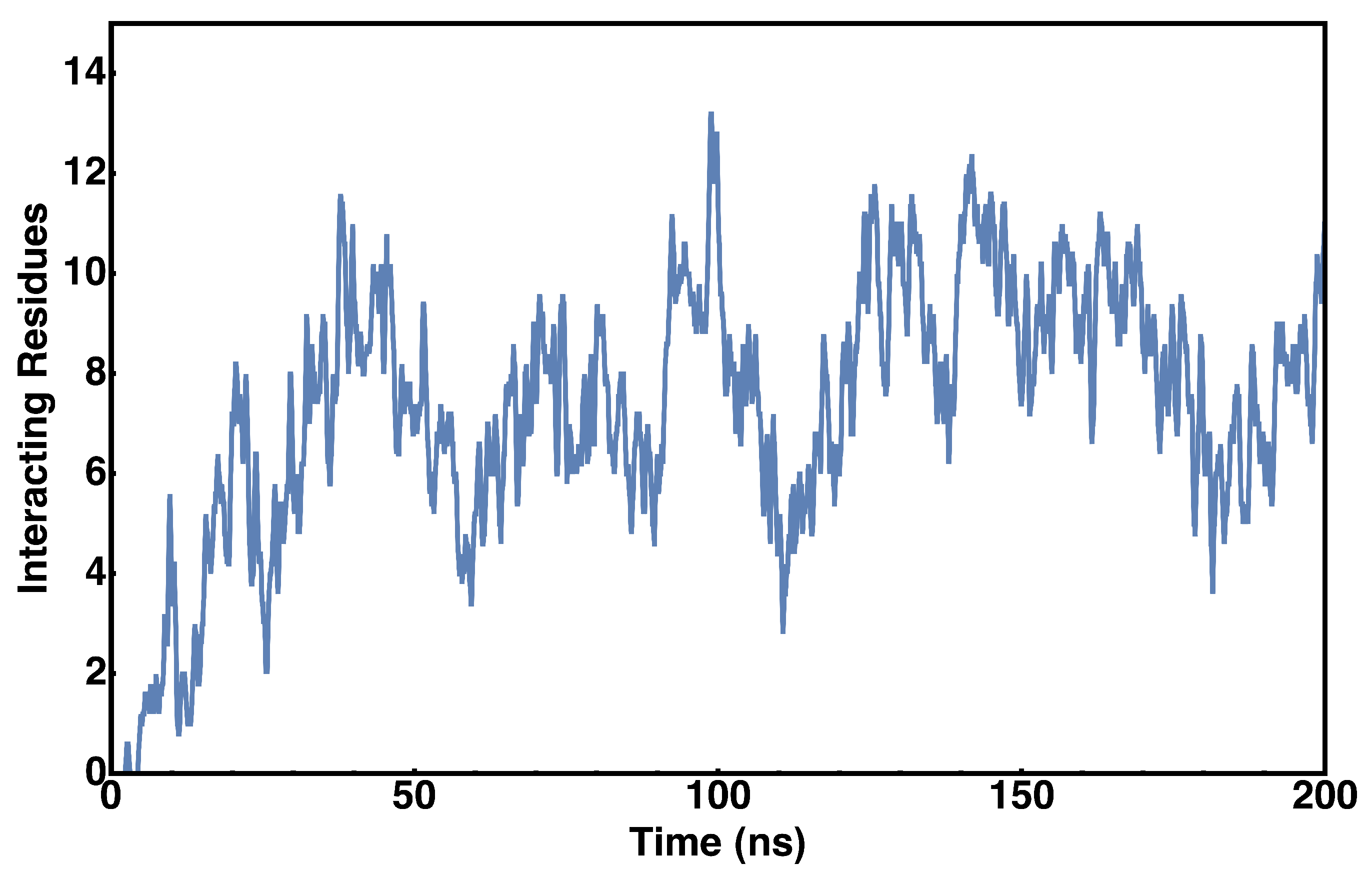
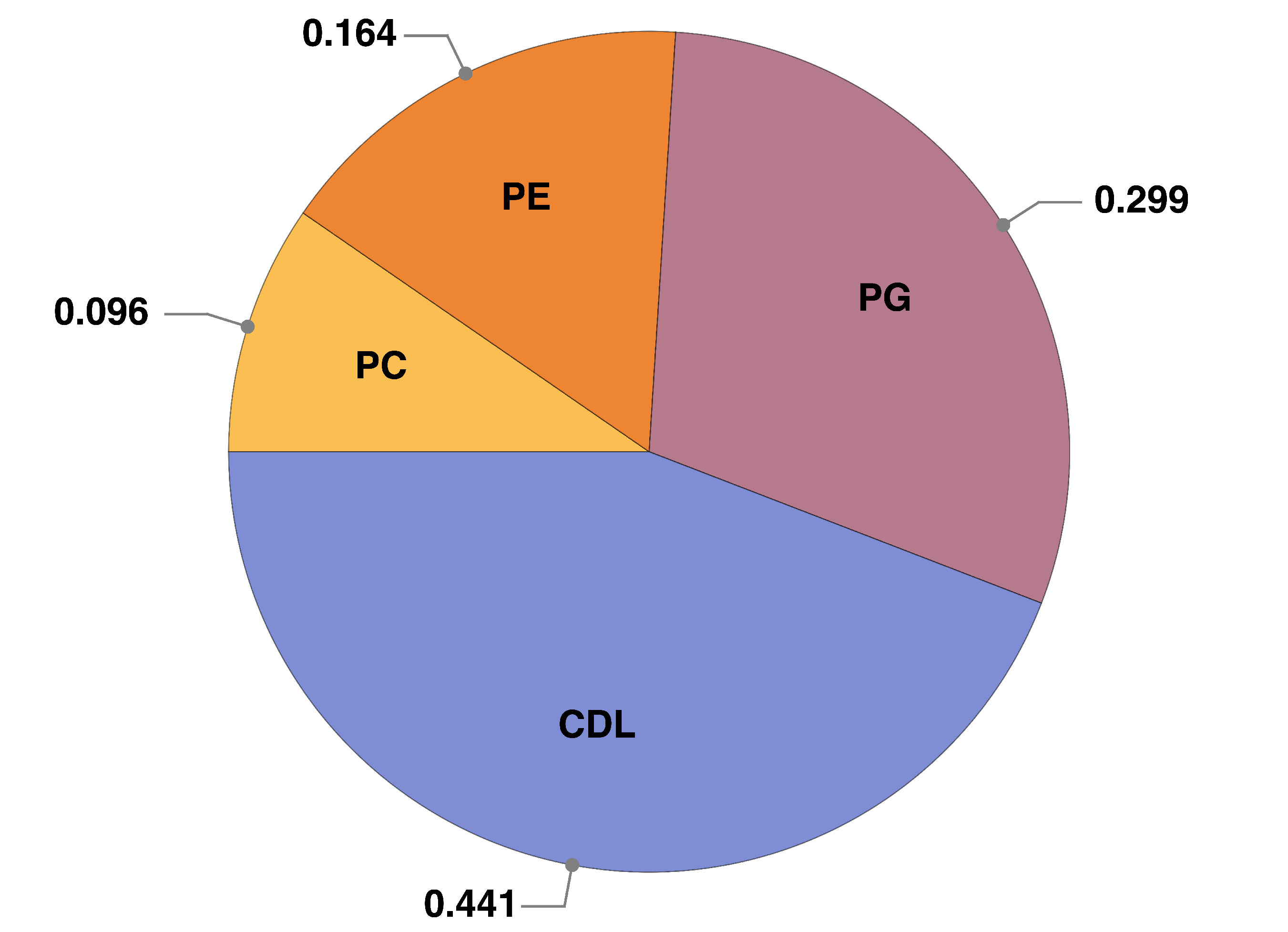
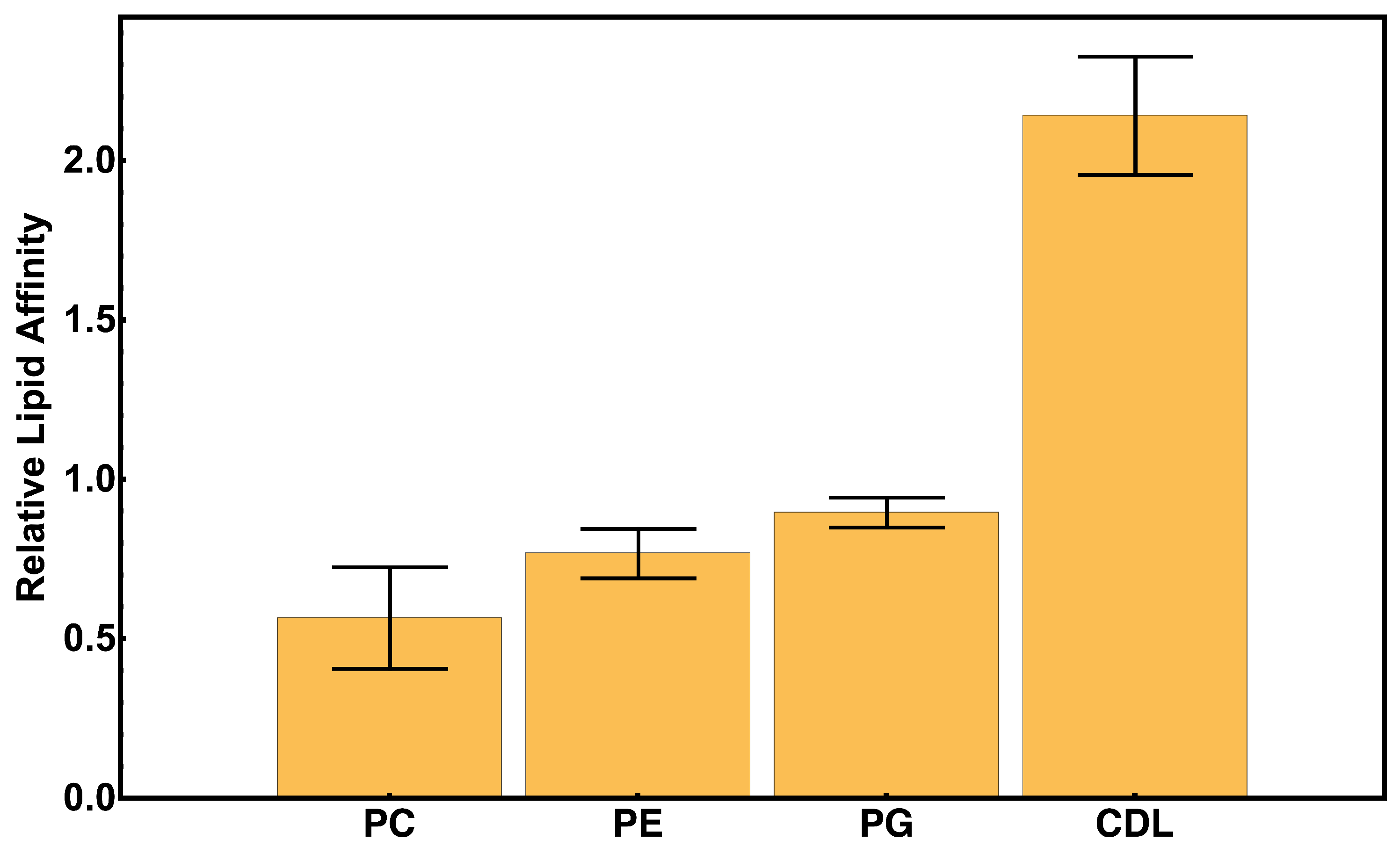
2.3. Protein–Lipid Interactions During cyt c2 Diffusion
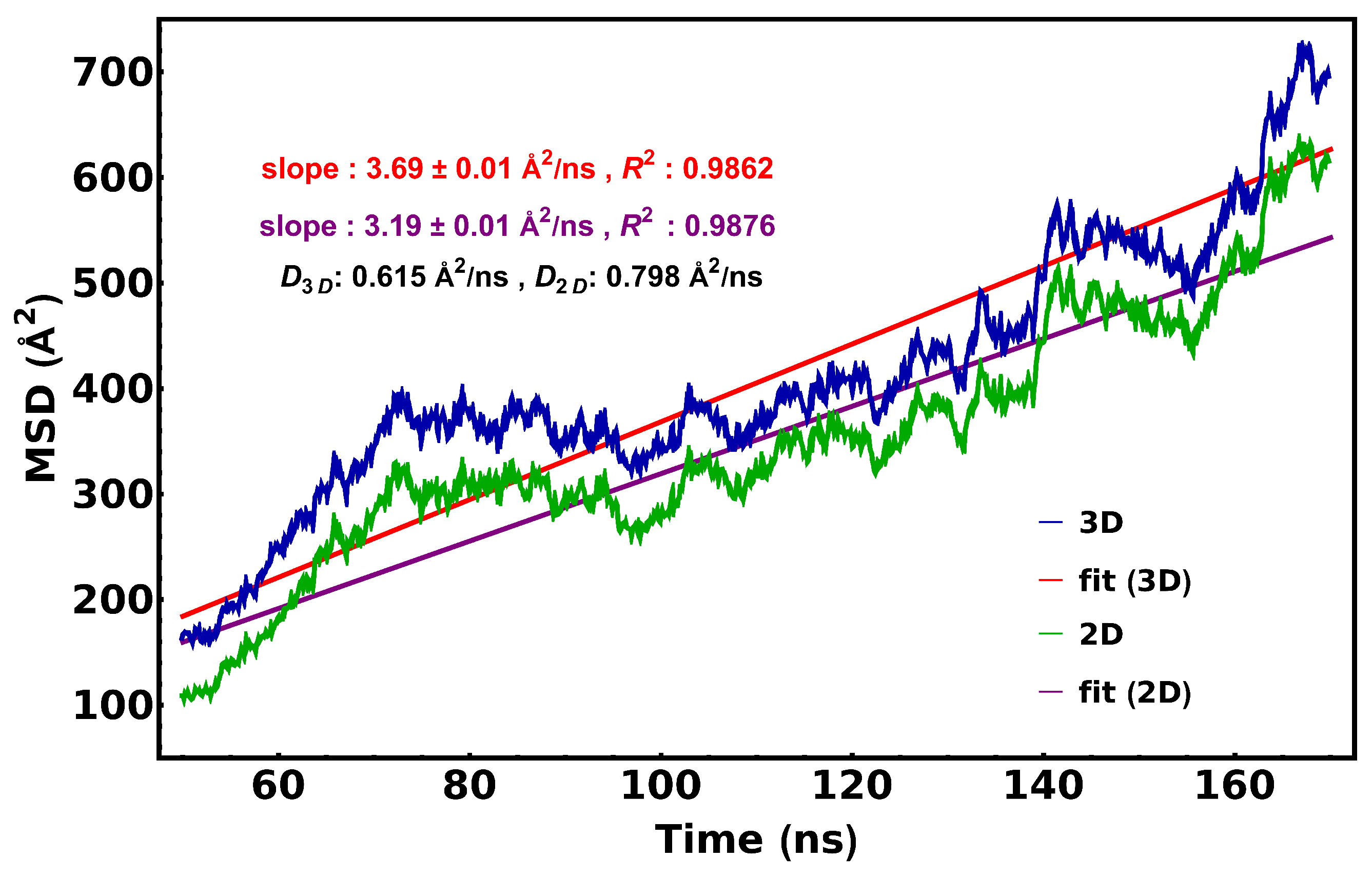
2.4. Calculation of cyt c2 Diffusion Coefficient
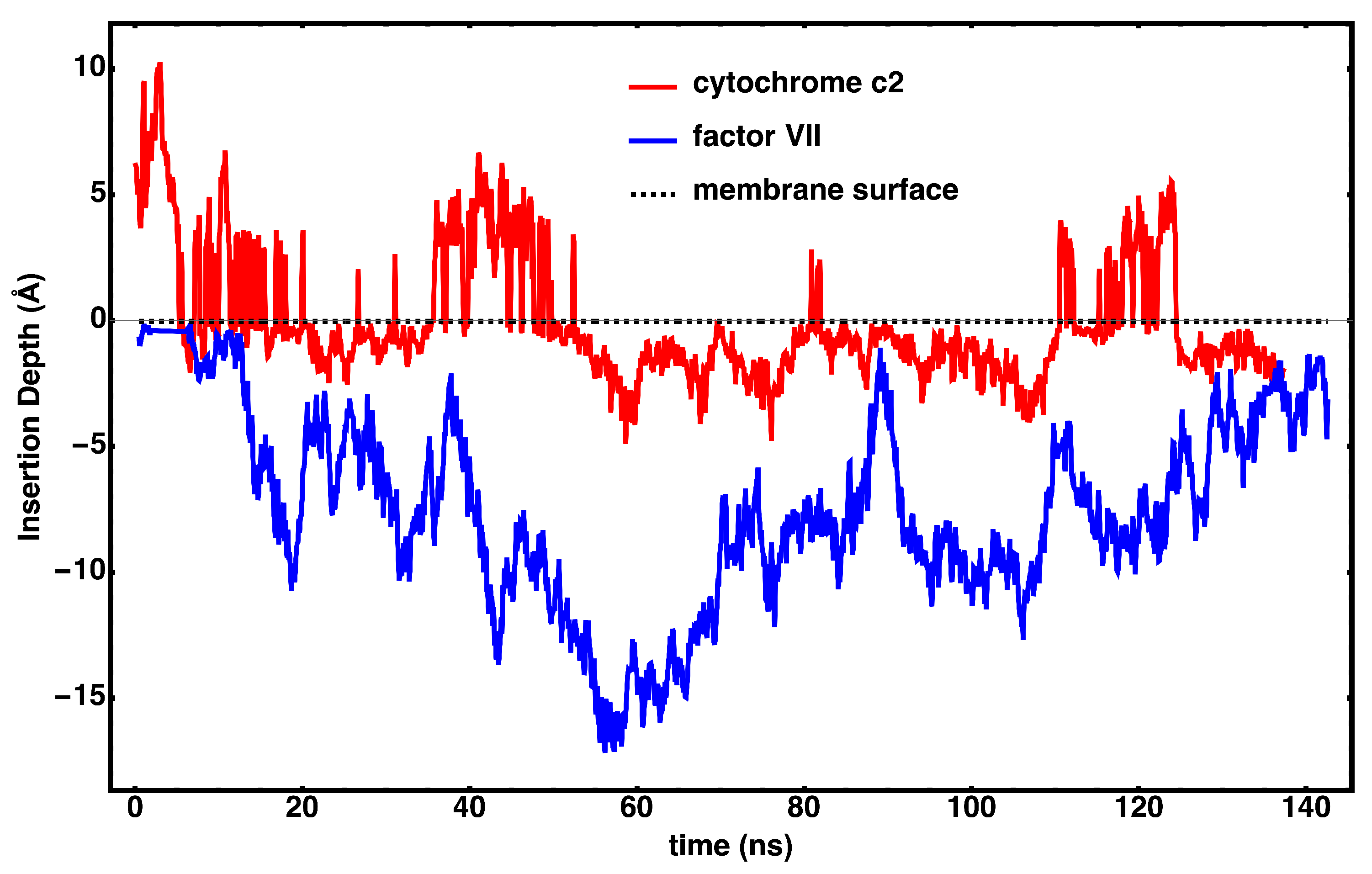
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Shimakawa, G. Electron transport in cyanobacterial thylakoid membranes: Are cyanobacteria simple models for photosynthetic organisms? J. Exp. Bot. 2023, 74, 3476–3487. [Google Scholar] [CrossRef] [PubMed]
- Sener, M.; Strumpfer, J.; Singharoy, A.; Hunter, C.N.; Schulten, K. Overall energy conversion efficiency of a photosynthetic vesicle. eLife 2016, 5, e09541. [Google Scholar] [CrossRef]
- Singharoy, A.; Barragan, A.M.; Thangapandian, S.; Tajkhorshid, E.; Schulten, K. Binding Site Recognition and Docking Dynamics of a Single Electron Transport Protein: Cytochrome c2. J. Am. Chem. Soc. 2016, 138, 12077–12089. [Google Scholar] [CrossRef]
- Woronowicz, K.; Harrold, J.W.; Kay, J.M.; Niederman, R.A. Structural and Functional Proteomics of Intracytoplasmic Membrane Assembly in Rhodobacter sphaeroides. J. Mol. Microbiol. Biotech. 2013, 23, 48–62. [Google Scholar] [CrossRef]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of Biological Electron-transfer. Nature 1992, 355, 796. [Google Scholar] [CrossRef]
- Moser, C.C.; Dutton, P.L. Cytochrome c and c2 binding dynamics and electron transfer with photosynthetic reaction center protein and other integral membrane redox proteins. Biochemistry 1988, 27, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Marcaida, M.J.; Schlarb-Ridley, B.G.; Worrall, J.A.; Wastl, J.; Evans, T.J.; Bendall, D.S.; Luisi, B.F.; Howe, C.J. Structure of Cytochrome c6A, a Novel Dithio-cytochrome of Arabidopsis thaliana, and its Reactivity with Plastocyanin: Implications for Function. J. Mol. Biol. 2006, 360, 968–977. [Google Scholar] [CrossRef]
- Dantas, J.M.; Campelo, L.M.; Duke, N.E.C.; Salgueiro, C.A.; Raj, P.P. The structure of PccH from Geobacter sulfurreducens, a novel low reduction potential monoheme cytochrome essential for accepting electrons from an electrode. FEBS J. 2015, 282, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.L.; Abresch, E.C.; Okamura, M.Y.; Yeh, A.P.; Ress, D.C.; Feher, G. X-ray Structure Determination of the Cytochrome C2: Reaction Center Electron Transfer Complex From Rhodobacter sphaeroides. J. Mol. Biol. 2002, 319, 501–515. [Google Scholar] [CrossRef]
- Stelter, M.; Melo, A.M.P.; Pereira, M.M.; Gomes, C.M.; Hreggvidsson, G.O.; Hjorleifsdottir, S.; Saraiva, L.M.; Teixeira, M.; Archer, M. A Novel Type of Monoheme Cytochrome c: Biochemical and Structural Characterization at 1.23 Å Resolution of Rhodothermus marinus Cytochrome c. Biochemistry 2008, 47, 11953–11963. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Saio, T.; Kumeta, H.; Uchida, T.; Inagaki, F.; Ishimori, K. Investigation of the redox-dependent modulation of structure and dynamics in human cytochrome c. Biochem. Biophys. Res. Commun. 2016, 469, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Benning, M.M.; Wesenberg, G.; Caffrey, M.S.; Bartsch, R.G.; Meyer, T.E.; Cusanovich, M.A.; Rayment, I.; Holden, H.M. Molecular structure of cytochrome c2 isolated from Rhodobacter capsulatus determined at 2.5 Åresolution. J. Mol. Biol. 1991, 220, 673–685. [Google Scholar] [CrossRef]
- Kerfeld, C.; Anwar, H.; Interrante, R.; Merchant, S.; Yeates, T. The Structure of Chloroplast Cytochrome C6 at 1.9 Å Resolution: Evidence for Functional Oligomerization. J. Mol. Biol. 1996, 250, 627. [Google Scholar]
- Can, M.; Krucinska, J.; Zoppellaro, G.; Andersen, N.H.; Wedekind, J.E.; Hersleth, H.P.; Andersson, K.K.; Bren, K.L. Structural characterization of nitrosomonas europaea cytochrome c-552 variants with marked differences in electronic structure. Chembiochem 2013, 14, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Nurit, M.; Jean, J.; Vivian, S.; Abel, M. High resolution X-ray crystallographic structure of bovine heart cytochrome c and its application to the design of an electron transfer biosensor. Proteins Struct. Funct. Bioinform. 2008, 70, 83–92. [Google Scholar] [CrossRef]
- Schlame, M. Thematic Review Series: Glycerolipids. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J. Lipid Res. 2008, 49, 1607–1620. [Google Scholar] [CrossRef]
- Renner, L.D.; Weibel, D.B. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 6264–6269. [Google Scholar] [CrossRef]
- Hryc, C.F.; Mallampalli, V.K.P.S.; Bovshik, E.I. Structural insights into cardiolipin replacement by phosphatidylglycerol in a cardiolipin-lacking yeast respiratory supercomplex. Nat. Commun. 2023, 14, 2783. [Google Scholar] [CrossRef]
- Overfield, R.E.; Wraight, C.A. Oxidation of cytochromes c and c2 by bacterial photosynthetic reaction centers in phospholipid vesicles. 1. Studies with neutral membranes. Biochemistry 1980, 19, 3322–3327. [Google Scholar] [CrossRef]
- Overfield, R.E.; Wraight, C.A. Oxidation of cytochromes c and c2 by bacterial photosynthetic reaction centers in phospholipid vesicles. 2. Studies with negative membranes. Biochemistry 1980, 19, 3328–3334. [Google Scholar] [CrossRef]
- Snozzi, M.; Crofts, A.R. Kinetics of the c-cytochromes in chromatophores from Rhodopseudomonas sphaeroides as a function of the concentration of cytochrome c2. Influence of this concentration on the oscillation of the secondary acceptor of the reaction centers QB. Biochim. Biophys. Acta Bioener. 1985, 809, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Singharoy, A.; Tajkhorshid, E. Anionic Lipids Confine Cytochrome c2 to the Surface of Bioenergetic Membranes Without Compromising Its Interaction with Redox Partners. Biochemistry 2022, 61, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, Y.Z.; Pogorelov, T.V.; Arcario, M.J.; Christensen, G.A.; Tajkhorshid, E. Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys. J. 2012, 102, 2130–2139. [Google Scholar] [CrossRef]
- Vermaas, J.V.; Pogorelov, T.V.; Tajkhorshid, E. Extension of the Highly Mobile Membrane Mimetic to Transmembrane Systems Through Customized in Silico Solvents. J. Phys. Chem. B 2017, 121, 3764–3776. [Google Scholar] [CrossRef] [PubMed]
- Lihan, M.; Tajkhorshid, E. Improved Highly Mobile Membrane Mimetic Model for Investigating Protein–Cholesterol Interactions. J. Chem. Inf. Model. 2024, 64, 4822–4834, Published. [Google Scholar] [CrossRef]
- Hasdemir, H.S.; Li, Y.; Kelich, P.; Wen, P.C.; Tajkhorshid, E. Characterization of membrane binding and protein-lipid interactions at atomic level with accelerated HMMM model. Methods Mol. Biol. 2025, in press. [Google Scholar]
- Russel, N.J.; Harwood, J.L. Changes in the Acyl Lipid Composition of Photosynthetic Bacteria Grown Under Photosynthetic and Non-Photosynthetic Conditions. Biophys. J. 1979, 181, 339–345. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-based Graphical User Interface for CHARMM. J. Comp. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Lamoureux, G.; Harder, E.; Vorobyov, I.V.; Roux, B.; MacKerell, A.D., Jr. A Polarizable Model of Water for Molecular Dynamics Simulations of Biomolecules. Chem. Phys. Lett. 2006, 418, 245–249. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD – Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D. CHARMM36 All-atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comp. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Autenrieth, F.; Tajkhorshid, E.; Schulten, K.; Luthey-Schulten, Z. Role of Water in Transient Cytochrome C2 Docking. J. Phys. Chem. B 2004, 108, 20376–20387. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant Pressure Molecular Dynamics Algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Muller, M.P.; Mortenson, A.; Sedzro, J.C.; Wen, P.C.; Morrissey, J.H.; Tajkhorshid, E. Membrane-bound model of the ternary complex between factor VIIa/tissue factor and factor X. Blood Adv. 2025, 9, 729–740. [Google Scholar] [CrossRef]
- Sharma, V.K.; Srinivasan, H.; Gupta, J.; Mitra, S. Lipid lateral diffusion: Mechanisms and modulators. Soft Mat. 2024, 20, 7763–7796. [Google Scholar] [CrossRef]
- Yoch, D.; Arnon, D.; Sweeney, W. Characterization of Two Soluble Ferredoxins as Distinct From Bound Iron-sulfur Proteins in the Photosynthetic Bacterium Rhodospirillum Rubrum. J. Biol. Chem. 1975, 250, 8330–8336. [Google Scholar] [CrossRef]
- Sanderson, D.; Anderson, L.; Gross, E. Determination of the Redox Potential and Diffusion Coefficient of the Protein Plastocyanin Using Optically Transparent Filar Electrodes. Biochim. Biophys. Acta Bioener. 1986, 852, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mejías, G.; Guerra-Castellano, A.; Díaz-Quintana, A.; De la Rosa, M.A.; Díaz-Moreno, I. Cytochrome c: Surfing Off of the Mitochondrial Membrane on the Tops of Complexes III and IV. Comput. Struct. Biotechnol. J. 2019, 17, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Solmaz, S.; Hunte, C. Structure of Complex III with Bound Cytochrome c in Reduced State and a Minimal Core Interface for Electron Transfer. J. Biol. Chem. 2008, 283, 17542–17549. [Google Scholar] [CrossRef]
- Capaldi, R.A.; Darley-Usmar, V.; Fuller, S.; Millett, F. Structural and functional features of the interaction of cytochrome c with complex III and cytochrome c oxidase. FEBS Lett. 1982, 138, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Northrup, S.H.; Reynolds, J.C.; Miller, C.M.; Forrest, K.J.; Boles, J.O. Diffusion-controlled association rate of cytochrome c and cytochrome c peroxidase in a simple electrostatic model. J. Am. Chem. Soc. 1986, 108, 8162–8170. [Google Scholar] [CrossRef]
- Brzezinski, P.; Moe, A.; Ädelroth, P. Structure and Mechanism of Respiratory III–IV Supercomplexes in Bioenergetic Membranes. Chem. Rev. 2021, 121, 9644–9673. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, A.; Tajkhorshid, E. Restricted Surface Diffusion of Cytochromes on Bioenergetic Membranes with Anionic Lipids. Membranes 2025, 15, 124. https://doi.org/10.3390/membranes15040124
Chan A, Tajkhorshid E. Restricted Surface Diffusion of Cytochromes on Bioenergetic Membranes with Anionic Lipids. Membranes. 2025; 15(4):124. https://doi.org/10.3390/membranes15040124
Chicago/Turabian StyleChan, Aaron, and Emad Tajkhorshid. 2025. "Restricted Surface Diffusion of Cytochromes on Bioenergetic Membranes with Anionic Lipids" Membranes 15, no. 4: 124. https://doi.org/10.3390/membranes15040124
APA StyleChan, A., & Tajkhorshid, E. (2025). Restricted Surface Diffusion of Cytochromes on Bioenergetic Membranes with Anionic Lipids. Membranes, 15(4), 124. https://doi.org/10.3390/membranes15040124