Diagnosis and Management of the Cryopyrin-Associated Periodic Syndromes (CAPS): What Do We Know Today?



Abstract
:1. Introduction
2. Epidemiology
3. Genetics
3.1. Frequent Variants of Uncertain Significance
3.2. Somatic Mutation/Somatic Mosaicism
4. Pathogenesis
5. Three Distinct Phenotypes Versus One Cryopyrin-Associated Periodic Syndromes (CAPS) Spectrum
6. Clinical Manifestations
6.1. Unspecific General Symptoms
6.2. Skin Manifestation
6.3. Musculoskeletal Involvement
6.4. Eye Involvement
6.5. Hearing Loss
6.6. Central Nervous Impairment
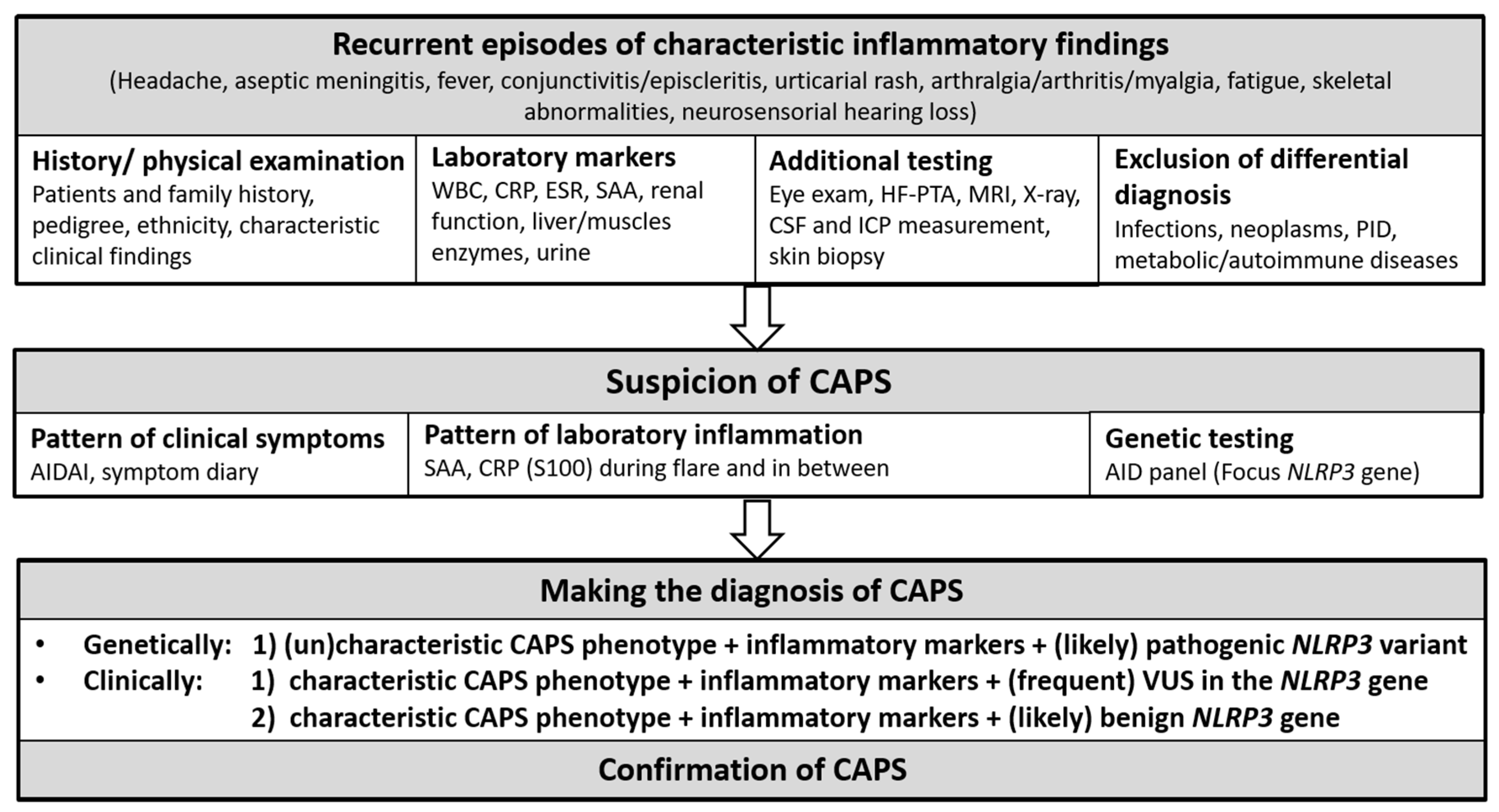
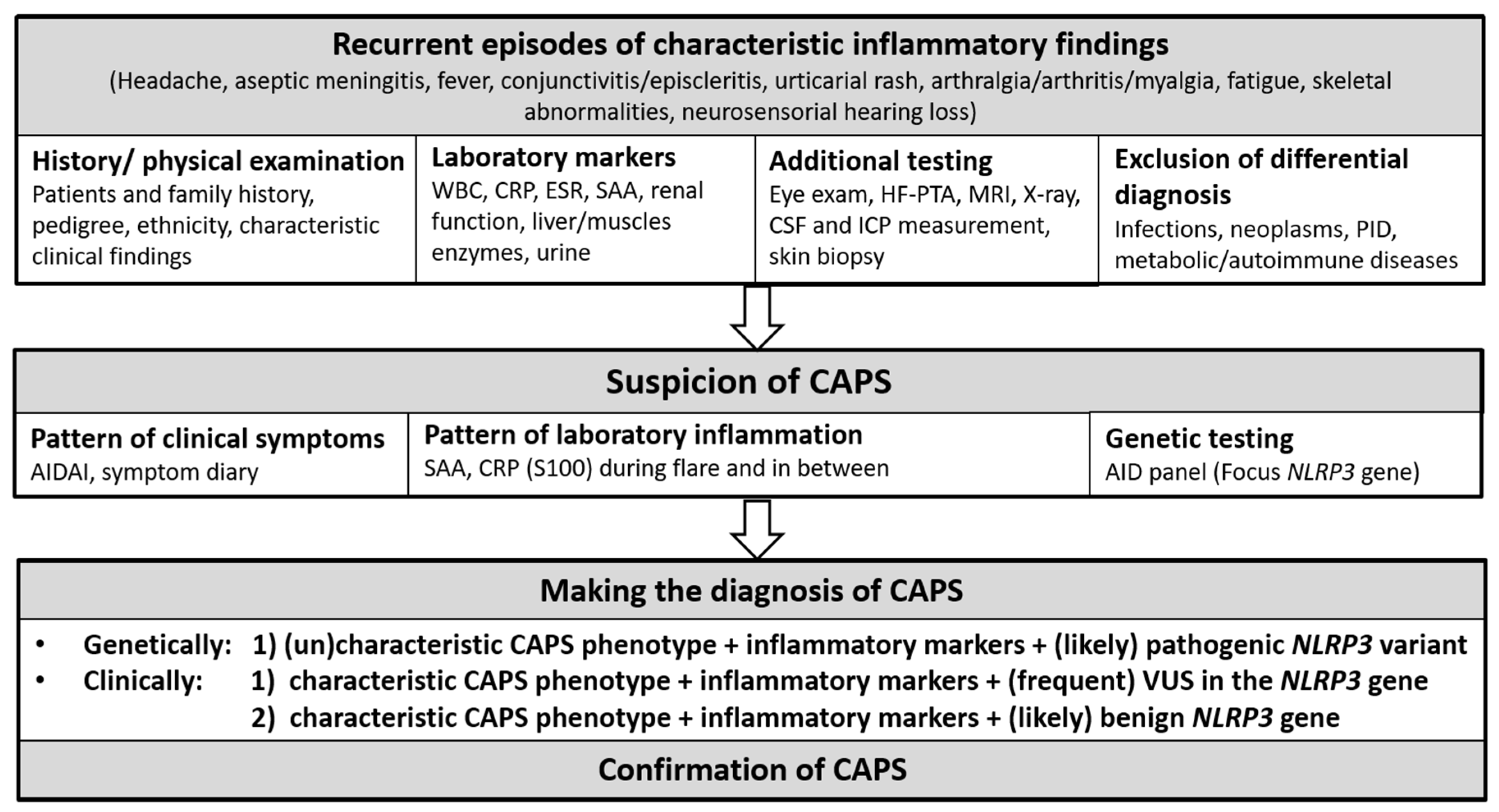
7. Diagnostic Approach
7.1. Diagnostic and Classification Criteria
7.1.1. Diagnostic Criteria
7.1.2. Classification Criteria
7.2. Diagnostic Challenges
8. Treatment
8.1. Anti-IL-1 Treatment
8.2. Supportive Therapy
8.3. Psychosocial Needs
8.4. Outlook Drug Development
9. Monitoring
10. Prognosis
Author Contributions
Funding
Conflicts of Interest
References
- Broderick, L. Hereditary Autoinflammatory Disorders: Recognition and Treatment. Immunol. Allergy Clin. N. Am. 2019, 39, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Broderick, L.; De Nardo, D.; Franklin, B.S.; Hoffman, H.M.; Latz, E. The Inflammasomes and Autoinflammatory Syndromes. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 395–424. [Google Scholar] [CrossRef] [PubMed]
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat. Genet. 1997, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997, 90, 797–807. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline Mutations in the Extracellular Domains of the 55 kDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Kastner, D.L. Autoinflammation: Past, Present, and Future. In Textbook of Autoinflammation; Hashkes, P.J., Laxer, R.M., Simon, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; Chapter 1; pp. 3–15. [Google Scholar]
- Drenth, J.P.; Cuisset, L.; Grateau, G.; Vasseur, C.; van de Velde-Visser, S.D.; de Jong, J.G.; Beckmann, J.S.; van der Meer, J.W.; Delpech, M. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat. Genet. 1999, 22, 178–181. [Google Scholar] [CrossRef]
- Houten, S.M.; Kuis, W.; Duran, M.; De Koning, T.J.; Van Royen-Kerkhof, A.; Romeijn, G.J.; Frenkel, J.; Dorland, L.; De Barse, M.M.; Huijbers, W.A.; et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 1999, 22, 175–177. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, J.; Prieur, A.-M.; Quartier, P.; Berquin, P.; Certain, S.; Cortis, E.; Teillac-Hamel, D.; Fischer, A.; Basile, G.D.S. Chronic Infantile Neurological Cutaneous and Articular Syndrome Is Caused by Mutations in CIAS1, a Gene Highly Expressed in Polymorphonuclear Cells and Chondrocytes. Am. J. Hum. Genet. 2002, 71, 198–203. [Google Scholar] [CrossRef] [Green Version]
- Aksentijevich, I.; Nowak, M.; Mallah, M.; Chae, J.J.; Watford, W.T.; Hofmann, S.R.; Stein, L.; Russo, R.; Goldsmith, D.; Dent, P.; et al. De novoCIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): A new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002, 46, 3340–3348. [Google Scholar] [CrossRef] [Green Version]
- Aksentijevich, I.; Putnam, C.D.; Remmers, E.F.; Mueller, J.L.; Le, J.; Kolodner, R.D.; Moak, Z.; Chuang, M.; Austin, F.; Goldbach-Mansky, R.; et al. The clinical continuum of cryopyrinopathies: Novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007, 56, 1273–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Chetrit, E.; Gattorno, M.; Gul, A.; Kastner, D.L.; Lachmann, H.J.; Touitou, I.; Ruperto, N. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): A Delphi study. Ann. Rheum. Dis. 2018, 77, 1558–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, H.M.; Kuemmerle-Deschner, J.B.; Goldbach-Mansky, R. Cryopyrine-Associated Periodic Syndromes (CAPS). In Textbook of Autoinflammation; Hashkes, P.J., Laxer, R.M., Simon, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; Chapter 19; pp. 347–365. [Google Scholar]
- Cuisset, L.; Jeru, I.; Dumont, B.; Fabre, A.; Cochet, E.; Le Bozec, J.; Delpech, M.; Amselem, S.; Touitou, I.; French CAPS study group. Mutations in the autoinflammatory cryopyrin-associated periodic syndrome gene: Epidemiological study and lessons from eight years of genetic analysis in France. Ann. Rheum. Dis. 2010, 70, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Lainka, E.; Neudorf, U.; Lohse, P.; Timmann, C.; Bielak, M.; Stojanov, S.; Huss, K.; Von Kries, R.; Niehues, T. Analysis of Cryopyrin-Associated Periodic Syndromes (CAPS) in German Children: Epidemiological, Clinical and Genetic Characteristics. Klin. Pädiatr. 2010, 222, 356–361. [Google Scholar] [CrossRef]
- Giat, E.; Lidar, M. Cryopyrin-associated periodic syndrome. Isr. Med. Assoc. J. 2014, 16, 659–661. [Google Scholar]
- Kummerle-Deschner, J.B. Cryopyrin-associated periodic syndrome. Z. Rheumatol. 2012, 71, 199–208. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Gregory, S.G.; Mueller, J.L.; Tresierras, M.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Fine structure mapping of CIAS1: Identification of an ancestral haplotype and a common FCAS mutation, L353P. Qual. Life Res. 2003, 112, 209–216. [Google Scholar] [CrossRef]
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Sarrauste de Menthière, C.; Terriere, S.; Pugnere, D.; Ruiz, M.; Demaille, J.; Touitou, I. INFEVERS: The Registry for FMF and hereditary inflammatory disorders mutations. Nucleic Acids Res. 2003, 31, 282–285. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B.; Verma, D.; Endres, T.; Broderick, L.; De Jesus, A.A.; Hofer, F.; Blank, N.; Krause, K.; Rietschel, C.; Horneff, G.; et al. Brief Report: Clinical and Molecular Phenotypes of Low-Penetrance Variants of NLRP3: Diagnostic and Therapeutic Challenges. Arthritis Rheumatol. 2017, 69, 2233–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoropoulou, K.; Wittkowski, H.; Busso, N.; Von Scheven-Gête, A.; Moix, I.; Vanoni, F.; Hengten, V.; Horneff, G.; Haas, J.-P.; Fischer, N.; et al. Increased Prevalence of NLRP3 Q703K Variant Among Patients with Autoinflammatory Diseases: An International Multicentric Study. Front. Immunol. 2020, 11, 877. [Google Scholar] [CrossRef] [PubMed]
- Shinar, Y.; Ceccherini, I.; Rowczenio, D.; Aksentijevich, I.; Arostegui, J.; Ben-Chétrit, E.; Boursier, G.; Gattorno, M.; Hayrapetyan, H.; Ida, H.; et al. ISSAID/EMQN Best Practice Guidelines for the Genetic Diagnosis of Monogenic Autoinflammatory Diseases in the Next-Generation Sequencing Era. Clin. Chem. 2020, 66, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Gavrilov, A.; Hofer, L.; Singh, A.; Öz, H.; Endres, T.; Schäfer, I.; Handgretinger, R.; Hartl, D.; Kuemmerle-Deschner, J. A functional inflammasome activation assay differentiates patients with pathogenic NLRP3 mutations and symptomatic patients with low penetrance variants. Clin. Immunol. 2015, 157, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Schuh, E.; Groß, C.J.; Wagner, D.; Schlüter, M.; Groß, O.; Kümpfel, T. MCC950 blocks enhanced interleukin-1beta production in patients with NLRP3 low penetrance variants. Clin. Immunol. 2019, 203, 45–52. [Google Scholar] [CrossRef]
- D’Osualdo, A.; Ferlito, F.; Prigione, I.; Obici, L.; Meini, A.; Zulian, F.; Pontillo, A.; Corona, F.; Barcellona, R.; Di Duca, M.; et al. Neutrophils from patients withTNFRSF1A mutations display resistance to tumor necrosis factor—Induced apoptosis: Pathogenetic and clinical implications. Arthritis Rheum. 2006, 54, 998–1008. [Google Scholar] [CrossRef]
- Touitou, I.; Askentijevich, I. Genetic Approach to the Diagnosis of Autoinflammatory Diseases. In Textbook of Autoinflammation; Hashkes, P.J., Laxer, R.M., Simon, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; Chapter 12; pp. 225–237. [Google Scholar]
- Hoffman, H.M.; Broderick, L. Editorial: It Just Takes One: Somatic Mosaicism in Autoinflammatory Disease. Arthritis Rheumatol. 2017, 69, 253–256. [Google Scholar] [CrossRef]
- Saito, M.; Fujisawa, A.; Nishikomori, R.; Kambe, N.; Nakata-Hizume, M.; Yoshimoto, M.; Ohmori, K.; Okafuji, I.; Yoshioka, T.; Kusunoki, T.; et al. Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2005, 52, 3579–3585. [Google Scholar] [CrossRef]
- Tanaka, N.; Izawa, K.; Saito, M.K.; Sakuma, M.; Oshima, K.; Ohara, O.; Nishikomori, R.; Morimoto, T.; Kambe, N.; Goldbach-Mansky, R.; et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: Results of an international multicenter collaborative study. Arthritis Rheum. 2011, 63, 3625–3632. [Google Scholar] [CrossRef] [Green Version]
- Labrousse, M.; Kevorkian-Verguet, C.; Boursier, G.; Rowczenio, D.; Maurier, F.; Lazaro, E.; Aggarwal, M.; Lemelle, I.; Mura, T.; Belot, A.; et al. Mosaicism in autoinflammatory diseases: Cryopyrin-associated periodic syndromes (CAPS) and beyond. A systematic review. Crit. Rev. Clin. Lab. Sci. 2018, 55, 432–442. [Google Scholar] [CrossRef]
- Nakagawa, K.; Gonzalez-Roca, E.; Souto, A.; Kawai, T.; Umebayashi, H.; Campistol, J.M.; Cañellas, J.; Takei, S.; Kobayashi, N.; Callejas-Rubio, J.L.; et al. SomaticNLRP3mosaicism in Muckle-Wells syndrome. A genetic mechanism shared by different phenotypes of cryopyrin-associated periodic syndromes. Ann. Rheum. Dis. 2013, 74, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mensa-Vilaro, A.; Bosque, M.T.; Magri, G.; Honda, Y.; Martínez-Banaclocha, H.; Casorran-Berges, M.; Sintes, J.; González-Roca, E.; Ruiz-Ortiz, E.; Heike, T.; et al. Brief Report: Late-Onset Cryopyrin-Associated Periodic Syndrome Due to Myeloid-Restricted Somatic NLRP3 Mosaicism. Arthritis Rheumatol. 2016, 68, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.; Gomes, S.M.; Aróstegui, J.I.; Mensa-Vilaro, A.; Omoyinmi, E.; Trojer, H.; Baginska, A.; Baroja-Mazo, A.; Pelegrin, P.; Savic, S.; et al. Late-Onset Cryopyrin-Associated Periodic Syndromes Caused by Somatic NLRP3 Mosaicism—UK Single Center Experience. Front. Immunol. 2017, 8, 1410. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Treviño, S.; González-Roca, E.; Ruiz-Ortiz, E.; Yagüe, J.; Ramos, E.; Aróstegui, J.I. First report of vertical transmission of a somatic NLRP3 mutation in cryopyrin-associated periodic syndromes: Table 1. Ann. Rheum. Dis. 2013, 72, 1109–1110. [Google Scholar] [CrossRef]
- Louvrier, C.; Assrawi, E.; El Khouri, E.; Melki, I.; Copin, B.; Bourrat, E.; Lachaume, N.; Cador-Rousseau, B.; Duquesnoy, P.; Piterboth, W.; et al. NLRP3-associated autoinflammatory diseases: Phenotypic and molecular characteristics of germline versus somatic mutations. J. Allergy Clin. Immunol. 2020, 145, 1254–1261. [Google Scholar] [CrossRef]
- Ye, Z.; Ting, J. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9. [Google Scholar] [CrossRef]
- Pétrilli, V.; Dostert, C.; Muruve, D.A.; Tschopp, J. The inflammasome: A danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007, 19, 615–622. [Google Scholar] [CrossRef]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Tran, T.A.T.; Grievink, H.W.; Lipinska, K.; Kluft, C.; Burggraaf, J.; Moerland, M.; Tasev, D.; Malone, K.E. Whole blood assay as a model for in vitro evaluation of inflammasome activation and subsequent caspase-mediated interleukin-1 beta release. PLoS ONE 2019, 14, e0214999. [Google Scholar] [CrossRef]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tassi, S.; Carta, S.; Delfino, L.; Caorsi, R.; Martini, A.; Gattorno, M.; Rubartelli, A. Altered redox state of monocytes from cryopyrin-associated periodic syndromes causes accelerated IL-1beta secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 9789–9794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosengren, S.; Mueller, J.L.; Anderson, J.P.; Niehaus, B.L.; Misaghi, A.; Anderson, S.; Boyle, D.L.; Hoffman, H.M. Monocytes from familial cold autoinflammatory syndrome patients are activated by mild hypothermia. J. Allergy Clin. Immunol. 2007, 119, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Kile, R.L.; Rusk, H.A. A Case of Cold Urticaria with an Unusual Family History. J. Am. Med. Assoc. 1940, 114, 1067–1068. [Google Scholar] [CrossRef]
- Muckle, T.J.; Wells, M. Urticaria, deafness, and amyloidosis: A new heredo-familial syndrome. Q. J. Med. 1962, 31, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Prieur, A.M.; Griscelli, C. Chronic meningo-cutaneo-articular syndrome in children. Rev. Rhum. Mal. Osteo-articul. 1980, 47, 645–649. [Google Scholar]
- Prieur, A.-M.; Griscelli, C. Arthropathy with rash, chronic meningitis, eye lesions, and mental retardation. J. Pediatr. 1981, 99, 79–83. [Google Scholar] [CrossRef]
- Ter Haar, N.; Oswald, M.; Jeyaratnam, J.; Anton, J.; Barron, K.; Brogan, P.; Cantarini, L.; Galeotti, C.; Grateau, G.; Hentgen, V.; et al. Recommendations for the management of autoinflammatory diseases. Pediatr. Rheumatol. 2015, 13, P133. [Google Scholar] [CrossRef] [Green Version]
- Caorsi, R.; Lepore, L.; Zulian, F.; Alessio, M.; Stabile, A.; Insalaco, A.; Finetti, M.; Battagliese, A.; Martini, G.; Bibalo, C.; et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis Res. Ther. 2013, 15, R33. [Google Scholar] [CrossRef] [Green Version]
- Kuemmerle-Deschner, J.B.; Hofer, F.; Endres, T.; Kortus-Goetze, B.; Blank, N.; Weißbarth-Riedel, E.; Schuetz, C.; Kallinich, T.; Krause, K.; Rietschel, C.; et al. Real-life effectiveness of canakinumab in cryopyrin-associated periodic syndrome. Rheumatology (Oxford) 2016, 55, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Neven, B.; Marvillet, I.; Terrada, C.; Ferster, A.; Boddaert, N.; Couloignier, V.; Pinto, G.; Pagnier, A.; Bodemer, C.; Bodaghi, B.; et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010, 62, 258–267. [Google Scholar] [CrossRef]
- Levy, R.; Gérard, L.; Kuemmerle-Deschner, J.; Lachmann, H.J.; Koné-Paut, I.; Cantarini, L.; Woo, P.; Naselli, A.; Bader-Meunier, B.; Insalaco, A.; et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: A series of 136 patients from the Eurofever Registry. Ann. Rheum. Dis. 2015, 74, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Neven, B.; Prieur, A.-M.; Maire, P.Q.D. Cryopyrinopathies: Update on pathogenesis and treatment. Nat. Clin. Pr. Rheumatol. 2008, 4, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Yadlapati, S.; Efthimiou, P. Impact of IL-1 inhibition on fatigue associated with autoinflammatory syndromes. Mod. Rheumatol. 2015, 26, 3–8. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B. CAPS—Pathogenesis, presentation and treatment of an autoinflammatory disease. Semin. Immunopathol. 2015, 37, 377–385. [Google Scholar] [CrossRef]
- Hoffman, D.H.M.; Wanderer, A.A.; Broide, D.H. Familial cold autoinflammatory syndrome: Phenotype and genotype of an autosomal dominant periodic fever. J. Allergy Clin. Immunol. 2001, 108, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathishkumar, D.; Al-Abadi, E.; Nicklaus-Wollenteit, I.; Moss, C.; Hawkins, P.N.; Gach, J.E. Early-onset urticaria: A marker of cryopyrin-associated periodic syndrome. Clin. Exp. Dermatol. 2017, 42, 579–581. [Google Scholar] [CrossRef]
- Herbert, V.; Ahmadi-Simab, K.; Reich, K.; Böer-Auer, A. Neutrophilic urticarial dermatosis (NUD) indicating Cryopyrin-associated periodic syndrome associated with a novel mutation of the NLRP3 gene. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 852–853. [Google Scholar] [CrossRef]
- Hill, S.C.; Namde, M.; Dwyer, A.; Poznanski, A.; Canna, S.; Goldbach-Mansky, R. Arthropathy of neonatal onset multisystem inflammatory disease (NOMID/CINCA). Pediatr. Radiol. 2007, 37, 145–152. [Google Scholar] [CrossRef]
- Finetti, M.; Omenetti, A.; Efederici, S.; Caorsi, R.; Gattorno, M. Chronic Infantile Neurological Cutaneous and Articular (CINCA) syndrome: A review. Orphanet J. Rare Dis. 2016, 11, 167. [Google Scholar] [CrossRef] [Green Version]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kim, H.J.; Brewer, C.; Zalewski, C.; Wiggs, E.; et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekic, S.; Yalcinbayir, O.; Kilic, S.S. Ocular Involvement in Muckle-Wells Syndrome. Ocul. Immunol. Inflamm. 2018, 28, 70–78. [Google Scholar] [CrossRef]
- Alejandre, N.; Ruiz-Palacios, A.; García-Aparicio, A.M.; Blanco-Kelly, F.; Bermúdez, S.; Fernández-Sanz, G.; Romero, F.I.; Aróstegui, J.I.; Ayuso, C.; Jiménez-Alfaro, I.; et al. Description of a new family with cryopyrin-associated periodic syndrome: Risk of visual loss in patients bearing the R260W mutation. Rheumatolog 2014, 53, 1095–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dollfus, H.; Häfner, R.; Hofmann, H.M.; Russo, R.A.G.; Denda, L.; Gonzales, L.D.; Decunto, C.; Premoli, J.; Melo-Gomez, J.; Jorge, J.P.; et al. Chronic Infantile Neurological Cutaneous and Articular/Neonatal Onset Multisystem Inflammatory Disease Syndrome Ocular Manifestations in a Recently Recognized Chronic Inflammatory Disease of Childhood. Arch. Ophthalmol. 2000, 118, 1386–1392. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, T.; Kawai, M.M.; Nishikomori, R.; Heike, T.; Takahashi, K. Obvious optic disc swelling in a patient with cryopyrin-associated periodic syndrome. Clin. Ophthalmol. 2013, 7, 1581–1585. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, N.; Brewer, C.C.; Zalewski, C.; King, K.A.; Butman, J.A.; Plass, N.; Henderson, C.; Goldbach-Mansky, R.; Kim, H.J. Cryopyrin-associated periodic syndromes: Otolaryngologic and audiologic manifestations. Otolaryngol. Head Neck Surg. 2011, 145, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Koitschev, A.; Gramlich, K.; Hansmann, S.; Benseler, S.; Plontke, S.K.; Koitschev, C.; Koetter, I.; Kuemmerle-Deschner, J.B. Progressive familial hearing loss in Muckle-Wells syndrome. Acta Oto-Laryngol. 2012, 132, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B.; Koitschev, A.; Tyrrell, P.N.; Plontke, S.K.; Deschner, N.; Hansmann, S.; Ummenhofer, K.; Lohse, P.; Koitschev, C.; Benseler, S.M. Early detection of sensorineural hearing loss in Muckle-Wells-syndrome. Pediatr. Rheumatol. 2015, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, H.; Prakash, P.; Ito, T.; Kim, H.J.; Brewer, C.C.; Harrow, D.; Roux, I.; Hosokawa, S.; Griffith, A.J. Genetic Hearing Loss Associated with Autoinflammation. Front. Neurol. 2020, 11, 141. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B.; Koitschev, A.; Ummenhofer, K.; Hansmann, S.; Plontke, S.K.; Koitschev, C.; Koetter, I.; Angermair, E.; Benseler, S.M. Hearing loss in Muckle-Wells syndrome. Arthritis Rheum. 2013, 65, 824–831. [Google Scholar] [CrossRef]
- Nakanishi, H.; Kawashima, Y.; Kurima, K.; Chae, J.J.; Ross, A.M.; Pinto-Patarroyo, G.; Patel, S.K.; Muskett, J.A.; Ratay, J.S.; Chattaraj, P.; et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E7766–E7775. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Wakiguchi, H.; Okazaki, F.; Nakamura, T.; Yasudo, H.; Kubo, M.; Sugahara, K.; Yamashita, H.; Suehiro, Y.; Okayama, N.; et al. Early canakinumab therapy for the sensorineural deafness in a family with Muckle-Wells syndrome due to a novel mutation of NLRP3 gene. Clin. Rheumatol. 2018, 38, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.K.; Horneff, G. Improvement of sensoneurinal hearing loss in a patient with Muckle-Wells syndrome treated with anakinra. Klin. Padiatr. 2010, 222, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Mamoudjy, N.; Maurey, H.; Marie, I.; Koné-Paut, I.; Deiva, K. Neurological outcome of patients with cryopyrin-associated periodic syndrome (CAPS). Orphanet J. Rare Dis. 2017, 12, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Parker, T.D.; Keddie, S.; Kidd, D.; Lane, T.; Maviki, M.; Hawkins, P.N.; Lachmann, H.J.; Ginsberg, L. Neurology of the cryopyrin-associated periodic fever syndrome. Eur. J. Neurol. 2016, 23, 1145–1151. [Google Scholar] [CrossRef] [Green Version]
- Toplak, N.; Frenkel, J.; Ozen, S.; Lachmann, H.J.; Woo, P.; Koné-Paut, I.; De Benedetti, F.; Neven, B.; Hofer, M.; Dolezalova, P.; et al. An International registry on Autoinflammatory diseases: The Eurofever experience. Ann. Rheum. Dis. 2012, 71, 1177–1182. [Google Scholar] [CrossRef]
- Mehr, S.; Allen, R.; Boros, C.; Adib, N.; Kakakios, A.; Turner, P.J.; Rogers, M.; Zurynski, Y.; Singh-Grewal, D. Cryopyrin-associated periodic syndrome in Australian children and adults: Epidemiological, clinical and treatment characteristics. J. Paediatr. Child. Health 2016, 52, 889–895. [Google Scholar] [CrossRef]
- Soon, G.S.; Laxer, R.M. Approach to recurrent fever in childhood. Can. Fam. Physician 2017, 63, 756–762. [Google Scholar]
- Georgin-Lavialle, S.; Fayand, A.; Rodrigues, F.; Bachmeyer, C.; Savey, L.; Grateau, G. Autoinflammatory diseases: State of the art. Presse Médicale 2019, 48, e25–e48. [Google Scholar] [CrossRef]
- Piram, M.; Koné-Paut, I.; Lachmann, H.J.; Frenkel, J.; Ozen, S.; Kuemmerle-Deschner, J.; Stojanov, S.; Simon, A.; Finetti, M.; Sormani, M.P.; et al. Validation of the Auto-Inflammatory Diseases Activity Index (AIDAI) for hereditary recurrent fever syndromes. Ann. Rheum. Dis. 2013, 73, 2168–2173. [Google Scholar] [CrossRef]
- Hawkins, P.; Lachmann, H.J.; Aganna, E.; McDermott, M.F. Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004, 50, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tan, X.; Zhang, J.; Li, S.; Mo, W.; Han, T.; Kuang, W.; Zhou, Y.; Deng, J. Gene mutations and clinical phenotypes in 15 Chinese children with cryopyrin-associated periodic syndrome (CAPS). Sci. China Life Sci. 2017, 60, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Wittkowski, H.; Kuemmerle-Deschner, J.B.; Austermann, J.; Holzinger, D.; Goldbach-Mansky, R.; Gramlich, K.; Lohse, P.; Jung, T.; Roth, J.; Benseler, S.M.; et al. MRP8 and MRP14, phagocyte-specific danger signals, are sensitive biomarkers of disease activity in cryopyrin-associated periodic syndromes. Ann. Rheum. Dis. 2011, 70, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, H.J.; Goodman, H.J.B.; Gilbertson, J.A.; Gallimore, J.R.; Sabin, C.A.; Gillmore, J.D.; Hawkins, P.N. Natural History and Outcome in Systemic AA Amyloidosis. N. Engl. J. Med. 2007, 356, 2361–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirmala, N.; Grom, A.; Gram, H. Biomarkers in systemic juvenile idiopathic arthritis: A comparison with biomarkers in cryopyrin-associated periodic syndromes. Curr. Opin. Rheumatol. 2014, 26, 543–552. [Google Scholar] [CrossRef]
- Serrano, M.; Ormazábal, A.; Anton, J.; Arostegui, J.I.; García-Cazorla, À. Cerebrospinal Fluid Neopterin and Cryopyrin-Associated Periodic Syndrome. Pediatr. Neurol. 2009, 41, 448–450. [Google Scholar] [CrossRef]
- Hashkes, P.J.; Barron, K.S.; Laxer, R.M. Clinical Approach to the Diagnosis of Autoinflammatory Diseases. In Textbook of Autoinflammation; Springer: Berlin/Heidelberg, Germany, 2019; pp. 203–223. [Google Scholar]
- Kuemmerle-Deschner, J.B.; Özen, S.; Tyrrell, P.N.; Kone-Paut, I.; Goldbach-Mansky, R.; Lachmann, H.; Blank, N.; Hoffman, H.M.; Weissbarth-Riedel, E.; Hügle, B.; et al. Diagnostic criteria for cryopyrin-associated periodic syndrome (CAPS). Ann. Rheum. Dis. 2017, 76, 942–947. [Google Scholar] [CrossRef]
- Gattorno, M.; Hofer, M.; Federici, S.; Vanoni, F.; Bovis, F.; Aksentijevich, I.; Anton, J.; Arostegui, J.I.; Barron, K.; Ben-Cherit, E.; et al. Classification criteria for autoinflammatory recurrent fevers. Ann. Rheum. Dis. 2019, 78, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Boursier, G.; Rittore, C.; Georgin-Lavialle, S.; Belot, A.; Galeotti, C.; Hachulla, E.; Hentgen, V.; Rossi-Semerano, L.; Sarrabay, G.; Touitou, A.I.; et al. Positive Impact of Expert Reference Center Validation on Performance of Next-Generation Sequencing for Genetic Diagnosis of Autoinflammatory Diseases. J. Clin. Med. 2019, 8, 1729. [Google Scholar] [CrossRef] [Green Version]
- Papa, R.; Rusmini, M.; Volpi, S.; Caorsi, R.; Picco, P.; Grossi, A.; Caroli, F.; Bovis, F.; Musso, V.; Obici, L.; et al. Next generation sequencing panel in undifferentiated autoinflammatory diseases identifies patients with colchicine-responder recurrent fevers. Rheumatology (Oxford) 2019, 59, 344–360. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B.; Welzel, T.; Hoertnagel, K.; Tsiflikas, I.; Hospach, A.; Liu, X.; Schlipf, S.; Hansmann, S.; Samba, S.D.; Griesinger, A.; et al. New variant in the IL1RN-gene (DIRA) associated with late-onset, CRMO-like presentation. Rheumatology 2020, 59, 3259–3263. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, S.; Lainka, E.; Horneff, G.; Holzinger, D.; Rieber, N.; Jansson, A.F.; Rösen-Wolff, A.; Erbis, G.; Prelog, M.; Brunner, J.; et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: A German PRO-KIND initiative. Pediatr. Rheumatol. 2020, 18, 1–11. [Google Scholar] [CrossRef]
- Smolen, J.S. Treat-to-target: Rationale and strategies. Clin. Exp. Rheumatol. 2012, 30 (Suppl. 73), S2–S6. [Google Scholar] [PubMed]
- Lepore, L.; Paloni, G.; Caorsi, R.; Alessio, M.; Rigante, D.; Ruperto, N.; Cattalini, M.; Tommasini, A.; Zulian, F.; Ventura, A.; et al. Follow-Up and Quality of Life of Patients with Cryopyrin-Associated Periodic Syndromes Treated with Anakinra. J. Pediatr. 2010, 157, 310–315.e1. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.H.; Plass, N.; Snow, J.; Wiggs, E.A.; Brewer, C.C.; King, K.A.; Zalewski, C.; Kim, H.J.; Bishop, R.; Hill, S.; et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: A cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012, 64, 2375–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle-Deschner, J.B.; Tyrrell, P.N.; Koetter, I.; Wittkowski, H.; Bialkowski, A.; Tzaribachev, N.; Lohse, P.; Koitchev, A.; Deuter, C.; Foell, D.; et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 2011, 63, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Kullenberg, T.; Löfqvist, M.; Leinonen, M.; Goldbach-Mansky, R.; Olivecrona, H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology 2016, 55, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Smith, J.; Lin, Y.; Tsai, W.L.; Kim, H.; Montealegre-Sanchez, G.; Chapelle, D.; Huang, Y.; Sibley, C.H.; Gadina, M.; Wesley, R.; et al. Cerebrospinal Fluid Cytokines Correlate with Aseptic Meningitis and Blood–Brain Barrier Function in Neonatal-Onset Multisystem Inflammatory Disease: Central Nervous System Biomarkers in Neonatal-Onset Multisystem Inflammatory Disease Correlate with Central Nervous System Inflammation. Arthritis Rheumatol. 2017, 69, 1325–1336. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, H.M.; Throne, M.L.; Amar, N.J.; Sebai, M.; Kivitz, A.J.; Kavanaugh, A.; Weinstein, S.P.; Belomestnov, P.; Yancopoulos, G.D.; Stahl, N.; et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: Results from two sequential placebo-controlled studies. Arthritis Rheum. 2008, 58, 2443–2452. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Throne, M.L.; Amar, N.J.; Cartwright, R.C.; Kivitz, A.J.; Soo, Y.; Weinstein, S.P. Long-Term Efficacy and Safety Profile of Rilonacept in the Treatment of Cryopryin-Associated Periodic Syndromes: Results of a 72-Week Open-Label Extension Study. Clin. Ther. 2012, 34, 2091–2103. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; Gitton, X.; Widmer, A.; Patel, N.; Hawkins, P.N. Use of Canakinumab in the Cryopyrin-Associated Periodic Syndrome. N. Engl. J. Med. 2009, 360, 2416–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle-Deschner, J.B.; Hachulla, E.; Cartwright, R.; Hawkins, P.N.; Tran, T.A.; Bader-Meunier, B.; Hoyer, J.; Gattorno, M.; Gul, A.; Smith, J.; et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann. Rheum. Dis. 2011, 70, 2095–2102. [Google Scholar] [CrossRef]
- Brogan, P.A.; Hofer, M.; Kuemmerle-Deschner, J.B.; Koné-Paut, I.; Roesler, J.; Kallinich, T.; Horneff, G.; Penadés, I.C.; Sevilla-Perez, B.; Goffin, L.; et al. Rapid and Sustained Long-Term Efficacy and Safety of Canakinumab in Patients with Cryopyrin-Associated Periodic Syndrome Ages Five Years and Younger. Arthritis Rheumatol. 2019, 71, 1955–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, R.A.G.; Melo-Gomes, S.; Lachmann, H.J.; Wynne, K.; Rajput, K.; Eleftheriou, D.; Edelsten, C.; Hawkins, P.N.; Brogan, P.A. Efficacy and safety of canakinumab therapy in paediatric patients with cryopyrin-associated periodic syndrome: A single-centre, real-world experience. Rheumatology 2014, 53, 665–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle-Deschner, J.B.; Ramos, E.; Blank, N.; Roesler, J.; Felix, S.D.; Jung, T.; Stricker, K.; Chakraborty, A.; Tannenbaum, S.; Wright, A.M.; et al. Canakinumab (ACZ885, a fully human IgG1 anti-IL-1beta mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res. Ther. 2011, 13, R34. [Google Scholar] [CrossRef] [Green Version]
- Sibley, C.H.; Chioato, A.; Felix, S.; Colin, L.; Chakraborty, A.; Plass, N.; Rodriguez-Smith, J.; Brewer, C.; King, K.; Zalewski, C.; et al. A 24-month open-label study of canakinumab in neonatal-onset multisystem inflammatory disease. Ann. Rheum. Dis. 2015, 74, 1714–1719. [Google Scholar] [CrossRef] [Green Version]
- Cipolletta, S.; Giudici, L.; Punzi, L.; Galozzi, P.; Sfriso, P. Health-related quality of life, illness perception, coping strategies and the distribution of dependency in autoinflammatory diseases. Clin. Exp. Rheumatol. 2019, 37, 156–157. [Google Scholar]
- Makay, B.; Emiroğlu, N.; Ünsal, E. Depression and anxiety in children and adolescents with familial Mediterranean fever. Clin. Rheumatol. 2009, 29, 375–379. [Google Scholar] [CrossRef]
- Giese, A.; Örnek, A.; Kılıç, L.; Kurucay, M.; Şendur, S.N.; Lainka, E.; Henning, B.F. Anxiety and depression in adult patients with familial Mediterranean fever: A study comparing patients living in Germany and Turkey. Int. J. Rheum. Dis. 2014, 20, 2093–2100. [Google Scholar] [CrossRef]
- Erbis, G.; Schmidt, K.; Hansmann, S.; Sergiichuk, T.; Michler, C.; Kuemmerle-Deschner, J.B.; Benseler, S. Living with autoinflammatory diseases: Identifying unmet needs of children, adolescents and adults. Pediatr. Rheumatol. 2018, 16, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; De Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrín, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Walle, L.V.; Stowe, I.B.; Šácha, P.; Lee, B.L.; Demon, D.; Fossoul, A.; Van Hauwermeiren, F.; Saavedra, P.H.; Šimon, P.; Šubrt, V.; et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Kümmerle-Deschner, J.B.; Tyrrell, P.N.; Reess, F.; Kotter, I.; Lohse, P.; Girschick, H.; Huemer, C.; Horneff, G.; Haas, J.-P.; Koitschev, A.; et al. Risk factors for severe Muckle-Wells syndrome. Arthritis Rheum. 2010, 62, 3783–3791. [Google Scholar] [CrossRef]
- Ter Haar, N.M.; Annink, K.V.; Al-Mayouf, S.M.; Amaryan, G.; Anton, J.; Barron, K.S.; Benseler, S.M.; Brogan, P.A.; Cantarini, L.; Cattalini, M.; et al. Development of the autoinflammatory disease damage index (ADDI). Ann. Rheum. Dis. 2016, 76, 821–830. [Google Scholar] [CrossRef]
- Ter Haar, N.M.; Van Delft, A.L.J.; Annink, K.V.; Van Stel, H.; Al-Mayouf, S.M.; Amaryan, G.; Anton, J.; Barron, K.; Benseler, S.; Brogan, P.A.; et al. In silico validation of the Autoinflammatory Disease Damage Index. Ann. Rheum. Dis. 2018, 77, 1599–1605. [Google Scholar] [CrossRef]
- Obici, L.; Merlini, G. Amyloidosis in autoinflammatory syndromes. Autoimmun. Rev. 2012, 12, 14–17. [Google Scholar] [CrossRef]

{kind=link}
| Clinical Manifestations and Characteristics of CAPS | |||
|---|---|---|---|
| Mild Phenotype (FCAS) | Moderate Phenotype (MWS) | Sever Phenotype (CINCA/NOMID) | |
| Disease onset | <6 months–adulthood | Early childhood–adulthood | Perinatal |
| Family history | Often positive | Often positive | Often negative (sporadic de novo mutations) |
| Inflammatory flares | Yes | Yes + continuous disease symptoms | Yes + continuous disease symptoms |
| Duration of inflammatory flares | 30 min–72 h | 1–3 Days ± subclinical | Persistent inflammation |
| Cold trigger | Yes | Possible | Rare |
| Dermatological manifestations | Cold-induced neutrophilic urticaria | Neutrophilic urticaria | Neutrophilic urticaria |
| Fever | 6–24 h after cold exposure possible | Particularly in childhood | Yes |
| Fatigue | Rare | Yes | Yes |
| Hearing loss | No | Yes | Yes |
| Ocular manifestation | Conjunctivitis | Conjunctivitis, episcleritis, optic disc edema/papilledema | Conjunctivitis, episcleritis, optic disc edema/papilledema |
| Muskulosceletal manifestations | Myalgia, arthralgia | Myalgia, arthralgia, oligoarthritis | Myalgia, arthralgia, (poly-) arthritis. epiphyseal bony overgrowth, limb-length discrepancies, contractures |
| Central nervous system manifestations | Headache | Headache, intermittent aseptic meningitis | Headache, chronic aseptic meningitis increased intracranial pressure, brain atrophy |
| Diagnostic Criteria for CAPS | |
|---|---|
| mandatory | + ≥2 of 6 clinical characteristic symptoms/signs |
| Raised inflammatory markers (C-reactive protein, serum amyloid A) |
|
| Eurofever/Printo Classification Criteria for CAPS | |
|---|---|
| Genetic Criteria | Clinical Criteria |
| Presence of a pathogenic/likely pathogenic NLRP3 gene variant | At least one among the following:
|
| Presence of a frequent NLRP3 gene variant of uncertain significance | At least two among the following:
|
| No presence of one of the above mentioned NLRP3 gene variants | At least two among the following:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Welzel, T.; Kuemmerle-Deschner, J.B. Diagnosis and Management of the Cryopyrin-Associated Periodic Syndromes (CAPS): What Do We Know Today? J. Clin. Med. 2021, 10, 128. https://doi.org/10.3390/jcm10010128
Welzel T, Kuemmerle-Deschner JB. Diagnosis and Management of the Cryopyrin-Associated Periodic Syndromes (CAPS): What Do We Know Today? Journal of Clinical Medicine. 2021; 10(1):128. https://doi.org/10.3390/jcm10010128
Chicago/Turabian StyleWelzel, Tatjana, and Jasmin B. Kuemmerle-Deschner. 2021. "Diagnosis and Management of the Cryopyrin-Associated Periodic Syndromes (CAPS): What Do We Know Today?" Journal of Clinical Medicine 10, no. 1: 128. https://doi.org/10.3390/jcm10010128
APA StyleWelzel, T., & Kuemmerle-Deschner, J. B. (2021). Diagnosis and Management of the Cryopyrin-Associated Periodic Syndromes (CAPS): What Do We Know Today? Journal of Clinical Medicine, 10(1), 128. https://doi.org/10.3390/jcm10010128