Cathepsin B Localizes in the Caveolae and Participates in the Proteolytic Cascade in Trabecular Meshwork Cells. Potential New Drug Target for the Treatment of Glaucoma

 ,
, 
Abstract
:1. Introduction
2. Experimental Section
2.1. Cell Culture
2.2. Preparation of Caveolae-Enriched Fractions
2.3. siRNA Transfection
2.4. Whole-Cell and Tissue Lysates Preparation
2.5. Western Blot Analysis
2.6. RNA Isolation and Quantitative Real-Time PCR
2.7. In Situ Zymography
2.8. Mechanical Stretch
2.9. ELISA
2.10. CTSBko Mice
2.11. IOP Measurements
2.12. Ex-Vivo Outflow Facility
2.13. Electron Microscopy (EM)
2.14. Statistical Analysis
3. Results
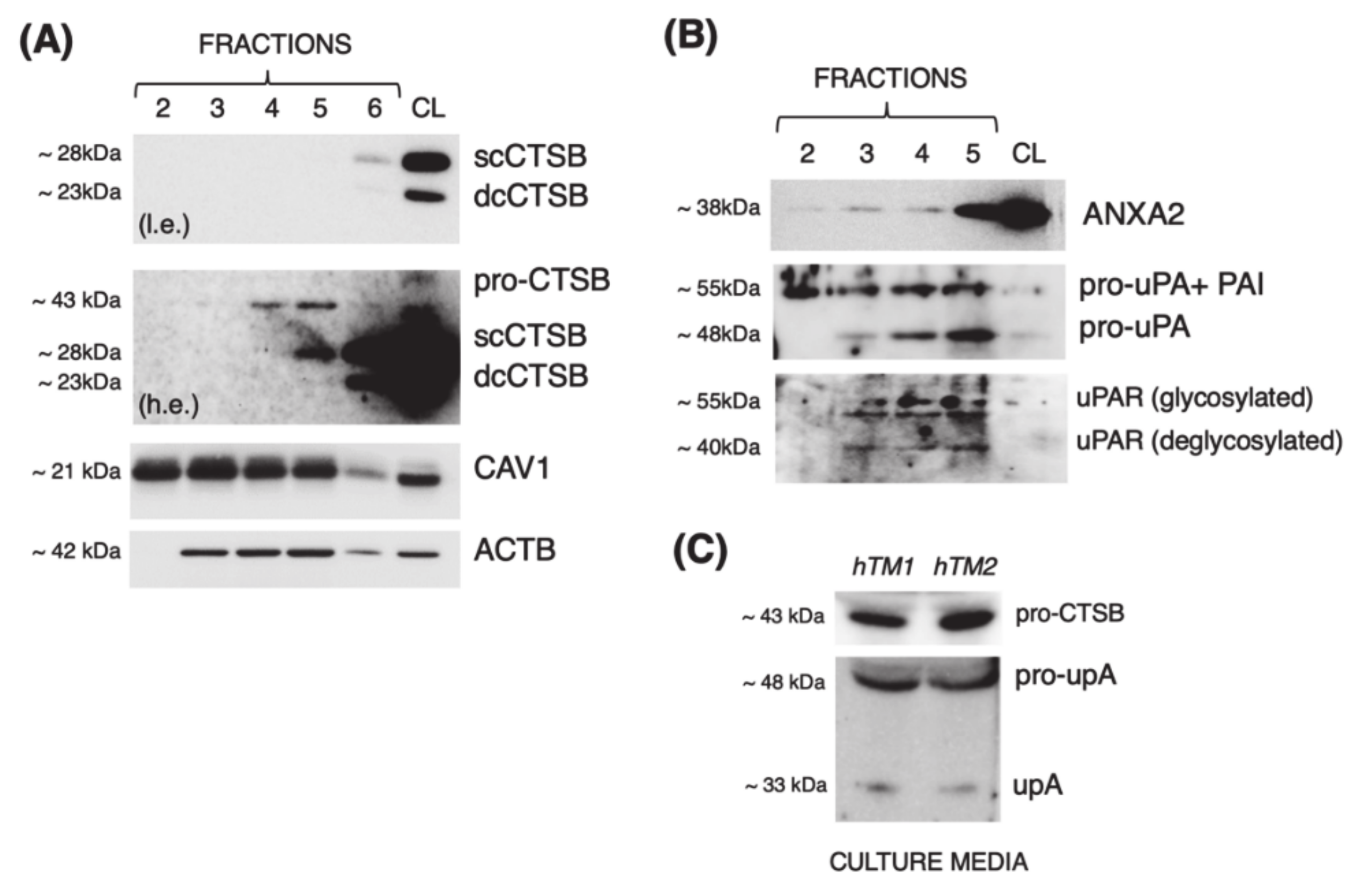
3.1. Cathepsin B Localizes in Caveolae with Components of the Proteolytic Cascade and Is Secreted into the Culture Media in Primary Cultures of TM Cells
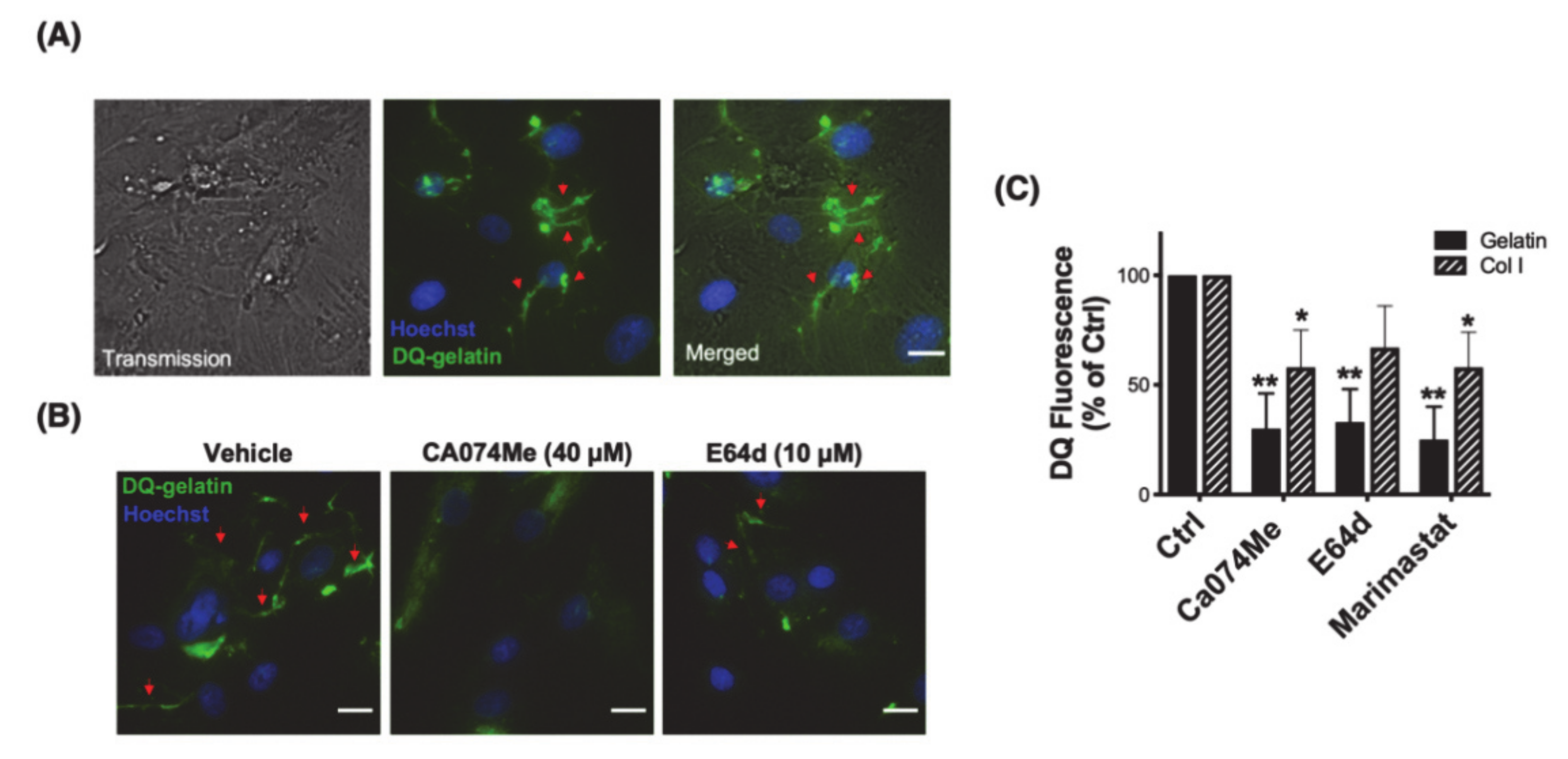
3.2. Cathepsin B Mediates Pericellular Proteolysis of ECM in TM Cells
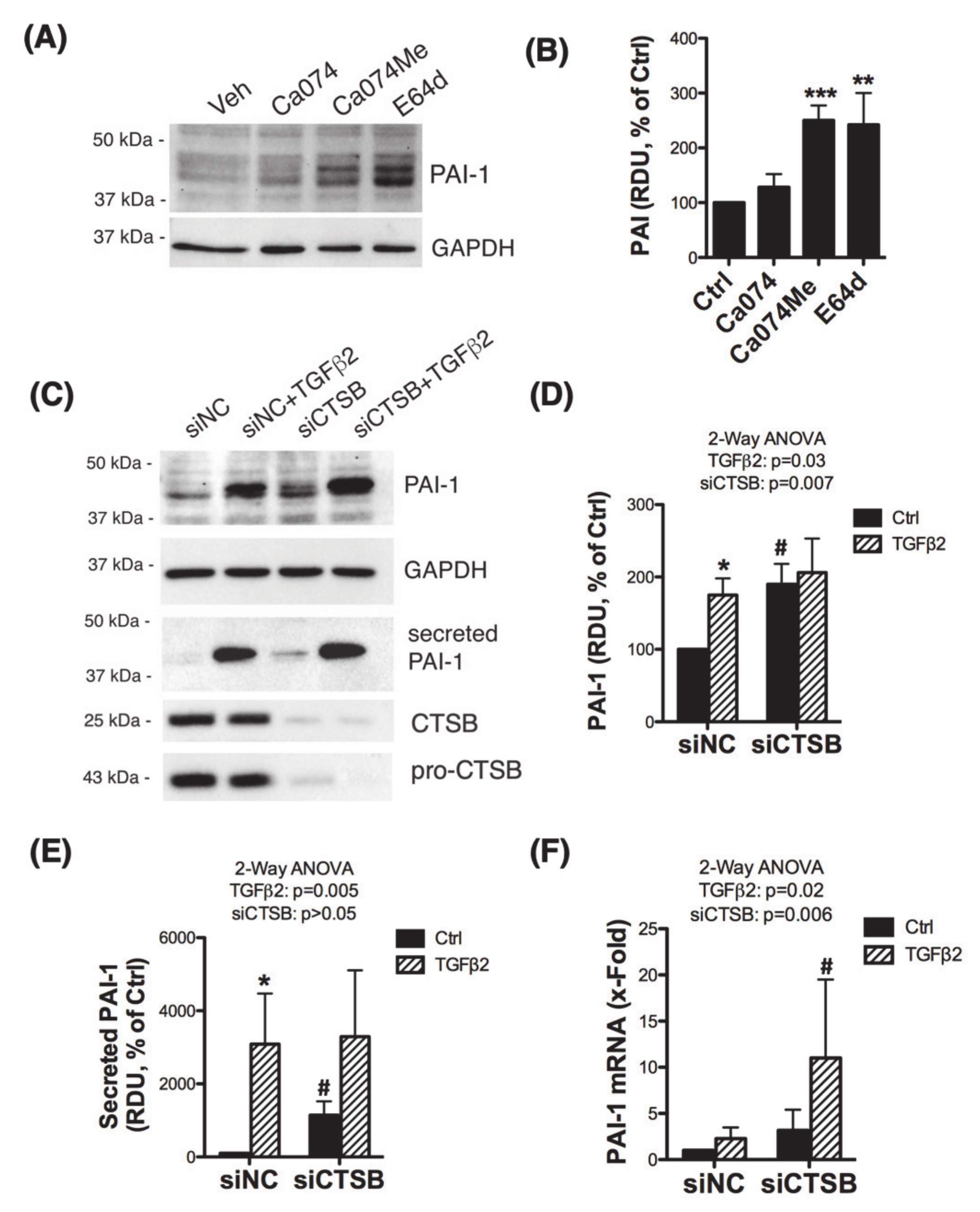
3.3. Inhibition of CTSB Upregulates and Increases the Secretion of Plasminogen Activator Inhibitor 1 (PAI-1)
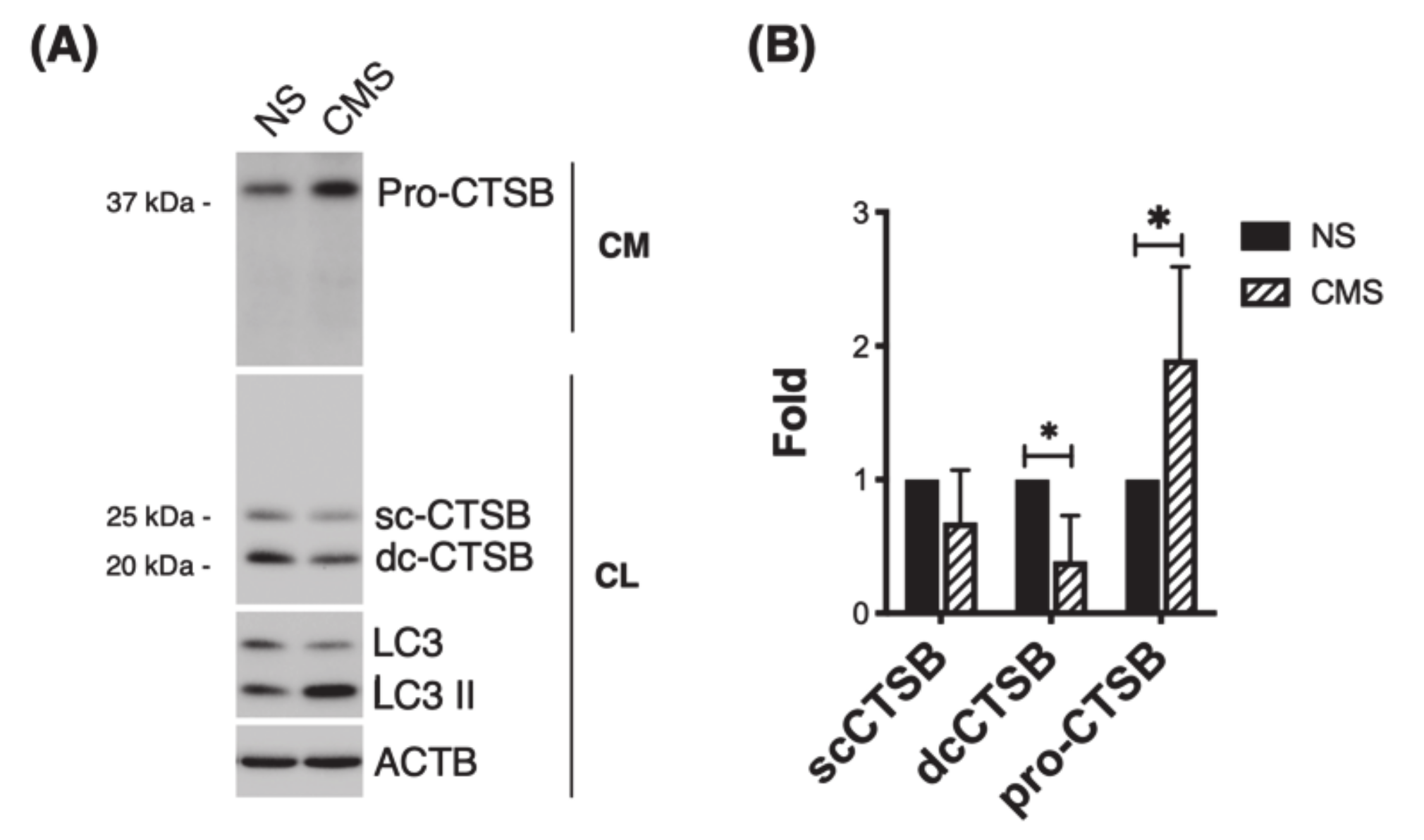
3.4. CTSB Is Secreted into the Culture Media in Response to Cyclic Mechanical Stretch
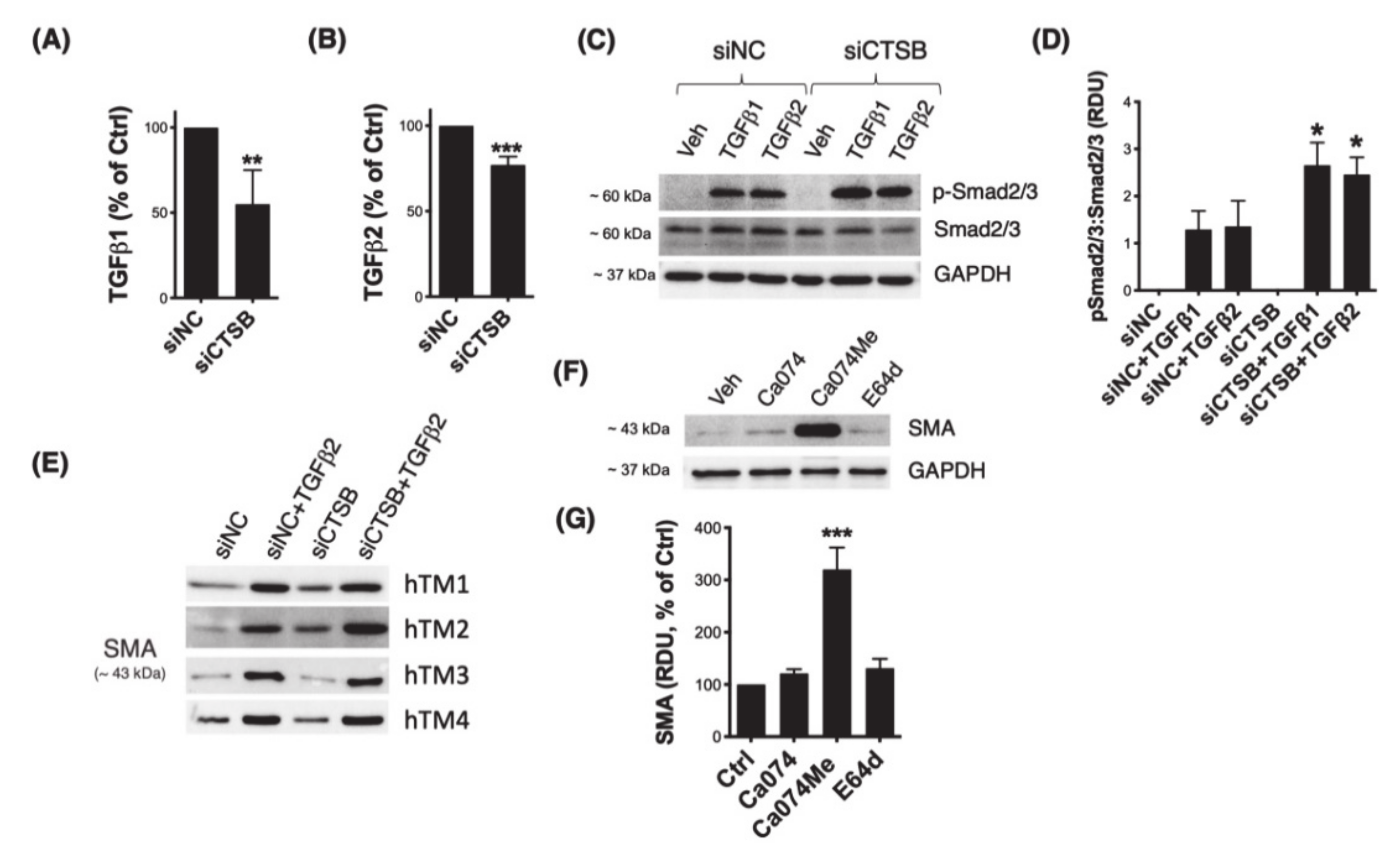
3.5. Downregulation of CTSB Decreases TGFβ1 and TGFβ2 and Modulates Smad2/3 Signaling Pathway
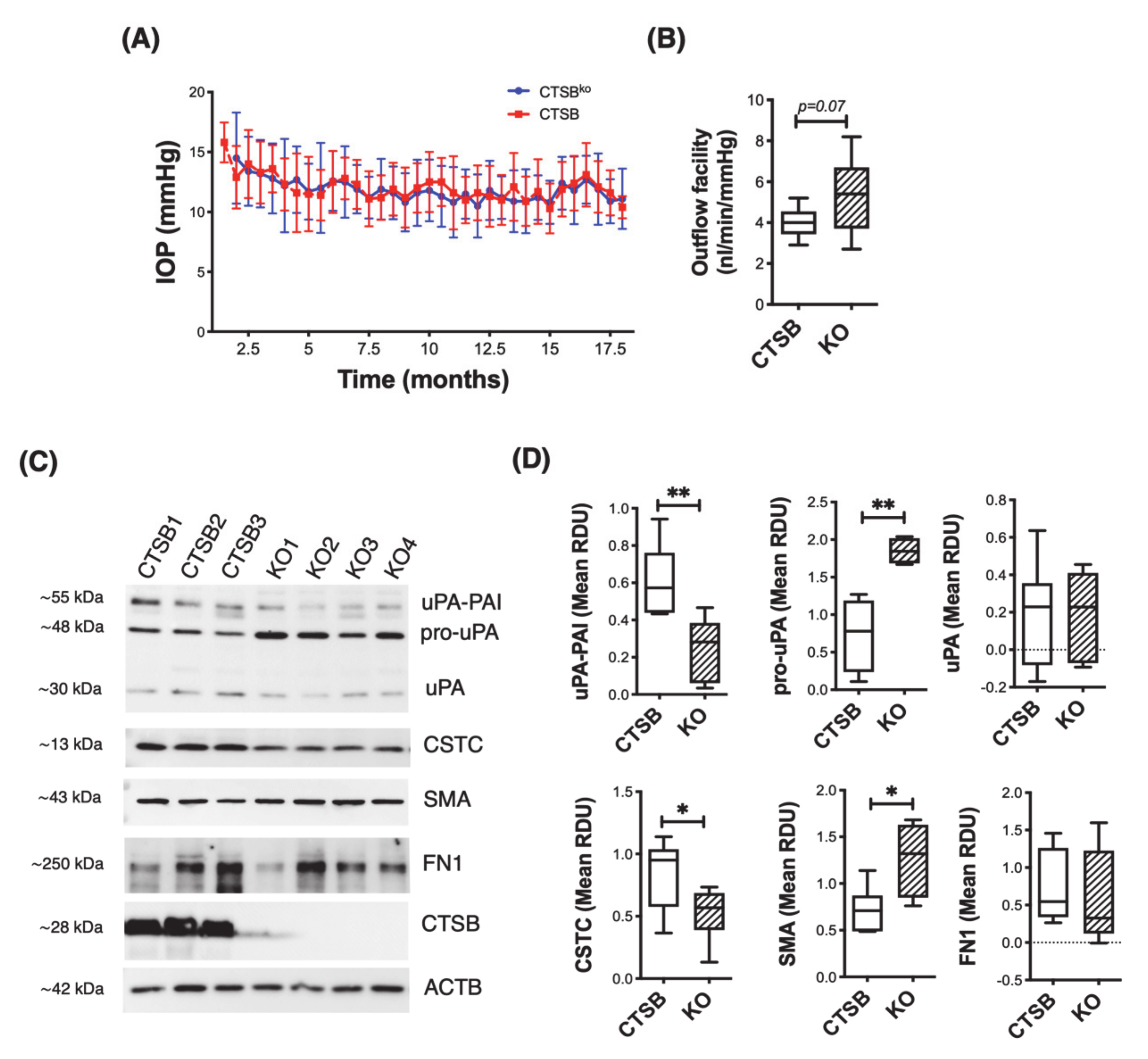
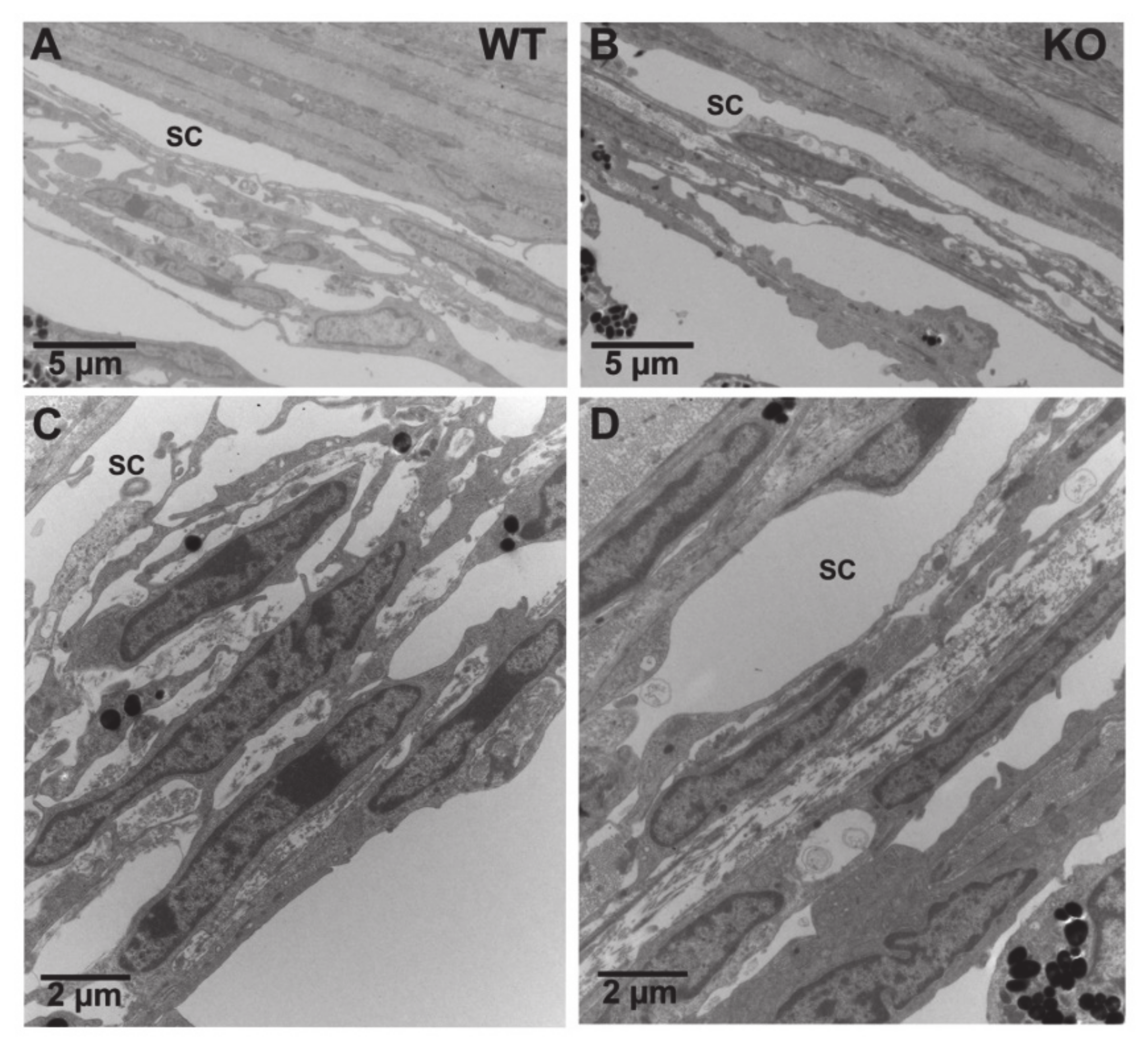
3.6. CTSB Knockout Mice Display Defective uPA Activation in the Iridocorneal Region
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bill, A.; Phillips, C.I. Uveoscleral drainage of aqueous humour in human eyes. Exp. Eye Res. 1971, 12, 275–281. [Google Scholar] [CrossRef]
- Quigley, H.A. Glaucoma. Lancet 2011, 377, 1367–1377. [Google Scholar] [CrossRef]
- Rohen, J.W.; Lütjen-Drecoll, E.; Flügel, C.; Meyer, M.; Grierson, I. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG). Exp. Eye Res. 1993, 56, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Acott, T.S.; Kelley, M.J. Extracellular matrix in the trabecular meshwork. Exp. Eye Res. 2008, 86, 543–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochiai, Y.; Ochiai, H. Higher concentration of transforming growth factor-beta in aqueous humor of glaucomatous eyes and diabetic eyes. Jpn. J. Ophthalmol. 2002, 46, 249–253. [Google Scholar] [CrossRef]
- Shepard, A.R.; Millar, J.C.; Pang, I.-H.; Jacobson, N.; Wang, W.-H.; Clark, A.F. Adenoviral gene transfer of active human transforming growth factor-{beta}2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2067–2076. [Google Scholar] [CrossRef] [Green Version]
- Junglas, B.; Kuespert, S.; Seleem, A.A.; Struller, T.; Ullmann, S.; Bösl, M.; Bosserhoff, A.; Köstler, J.; Wagner, R.; Tamm, E.R.; et al. Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. Am. J. Pathol. 2012, 180, 2386–2403. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.M.; Vranka, J.; Colvis, C.M.; Conger, D.M.; Alexander, J.P.; Fisk, A.S.; Samples, J.R.; Acott, T.S. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2649–2658. [Google Scholar]
- Spiga, M.-G.; Borrás, T. Development of a gene therapy virus with a glucocorticoid-inducible MMP1 for the treatment of steroid glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3029–3041. [Google Scholar] [CrossRef]
- Stamer, W.D.; Acott, T.S. Current understanding of conventional outflow dysfunction in glaucoma. Curr. Opin. Ophthalmol. 2012, 23, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Broemme, D.; Wilson, S. Role of Cysteine Cathepsins in Extracellular Proteolysis. In The Extracellular Matrix: An Overview, 1st ed.; Parks, W.C., Mecham, R.P., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 23–51. [Google Scholar]
- Porter, K.; Lin, Y.; Liton, P.B. Cathepsin B is up-regulated and mediates extracellular matrix degradation in trabecular meshwork cells following phagocytic challenge. PLoS ONE 2013, 8, e68668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Vreemann, A.; Qu, H.; Mayer, K.; Andersen, L.B.; Stefana, M.I.; Wehner, S.; Lysson, M.; Farcas, A.M.; Peters, C.; Reinheckel, T.; et al. Cathepsin B release from rodent intestine mucosa due to mechanical injury results in extracellular matrix damage in early post-traumatic phases. Biol. Chem. 2009, 390, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Jane, D.T.; Morvay, L.; Dasilva, L.; Cavallo-Medved, D.; Sloane, B.F.; Dufresne, M.J. Cathepsin B localizes to plasma membrane caveolae of differentiating myoblasts and is secreted in an active form at physiological pH. Biol. Chem. 2006, 387, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo-Medved, D.; Dosescu, J.; Linebaugh, B.E.; Sameni, M.; Rudy, D.; Sloane, B.F. Mutant K-ras regulates cathepsin B localization on the surface of human colorectal carcinoma cells. Neoplasia 2003, 5, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Cavallo-Medved, D.; Rudy, D.; Blum, G.; Bogyo, M.; Caglic, D.; Sloane, B.F. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp. Cell Res. 2009, 315, 1234–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallo-Medved, D.; Mai, J.; Dosescu, J.; Sameni, M.; Sloane, B.F. Caveolin-1 mediates the expression and localization of cathepsin, B.; pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J. Cell Sci. 2005, 118, 1493–1503. [Google Scholar] [CrossRef] [Green Version]
- Mitrović, A.; Pečar Fonović, U.; Kos, J. Cysteine cathepsins B and X promote epithelial-mesenchymal transition of tumor cells. Eur. J. Cell Biol. 2017, 96, 622–631. [Google Scholar] [CrossRef]
- Nettesheim, A.; Shim, M.S.; Hirt, J.; Liton, P.B. Transcriptome analysis reveals autophagy as regulator of TGFβ/Smad-induced fibrogenesis in trabecular meshwork cells. Sci. Rep. 2019, 9, 16092. [Google Scholar] [CrossRef]
- Keller, K.E.; Bhattacharya, S.K.; Borrás, T.; Brunner, T.M.; Chansangpetch, S.; Clark, A.F.; Dismuke, W.M.; Du, Y.; Elliott, M.H.; Ethier, C.R.; et al. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp. Eye Res. 2018, 171, 164–173. [Google Scholar] [CrossRef]
- Shim, M.S.; Nettesheim, A.; Hirt, J.; Liton, P.B. The Autophagic Protein LC3 Translocates to the Nucleus and Localizes in the Nucleolus Associated to NUFIP1 in Response to Cyclic Mechanical Stress. Autophagy; Taylor & Francis: Abingdon, UK, 2019; Volume 40, pp. 1–14. [Google Scholar] [CrossRef]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-Induced Systemic Cathepsin B Secretion Is Associated with Memory Function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deussing, J.; Roth, W.; Saftig, P.; Peters, C.; Ploegh, H.L.; Villadangos, J.A. Cathepsins B and D are dispensable for major histocompatibility complex class II-mediated antigen presentation. Proc. Natl. Acad. Sci. USA 1998, 95, 4516–4521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherwood, J.M.; Reina-Torres, E.; Bertrand, J.A.; Rowe, B.; Overby, D.R. Measurement of Outflow Facility Using iPerfusion. PLoS ONE 2016, 11, e0150694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, J.-Y.; Lamer, S.; Schümann, M.; Schmidt, M.R.; Krause, E.; Haucke, V. Quantitative proteomics analysis of detergent-resistant membranes from chemical synapses: Evidence for cholesterol as spatial organizer of synaptic vesicle cycling. Mol. Cell. Proteom. 2006, 5, 2060–2071. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Jevnikar, Z.; Doljak, B.; Kos, J. Role of cysteine cathepsins in matrix degradation and cell signalling. Connect. Tissue Res. 2008, 49, 193–196. [Google Scholar] [CrossRef]
- Sloane, B.F.; Yan, S.; Podgorski, I.; Linebaugh, B.E.; Cher, M.L.; Mai, J.; Cavallo-Medved, D.; Sameni, M.; Dosescu, J.; Moin, K. Cathepsin B and tumor proteolysis: Contribution of the tumor microenvironment. Semin. Cancer Biol. 2005, 15, 149–157. [Google Scholar] [CrossRef]
- Sameni, M.; Moin, K.; Sloane, B.F. Imaging proteolysis by living human breast cancer cells. Neoplasia 2000, 2, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Tu, C.; Ortega-Cava, C.F.; Chen, G.; Fernandes, N.D.; Cavallo-Medved, D.; Sloane, B.F.; Band, V.; Band, H. Lysosomal cathepsin B participates in the podosome-mediated extracellular matrix degradation and invasion via secreted lysosomes in v-Src fibroblasts. Cancer Res. 2008, 68, 9147–9156. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Moniwa, N.; Sugimura, M.; Shinohara, H.; Ohi, H.; Terao, T. Effects of membrane-associated cathepsin B on the activation of receptor-bound prourokinase and subsequent invasion of reconstituted basement membranes. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 1993, 1178, 55–62. [Google Scholar] [CrossRef]
- WuDunn, D. Mechanobiology of trabecular meshwork cells. Exp. Eye Res. 2009, 88, 718–723. [Google Scholar] [CrossRef]
- Liton, P.B.; Gonzalez, P. Stress response of the trabecular meshwork. J. Glaucoma 2008, 17, 378–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhou, Y.; Yu, X.; Huang, Q.; Fang, W.; Li, J.; Bonventre, J.V.; Sukhova, G.K.; Libby, P.; Shi, G.P. Differential Roles of Cysteinyl Cathepsins in TGF-β Signaling and Tissue Fibrosis. IScience 2019, 19, 607–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasabova, M.; Joulin-Giet, A.; Lecaille, F.; Gilmore, B.F.; Marchand-Adam, S.; Saidi, A.; Lalmanach, G. Regulation of TGF-β1-Driven Differentiation of Human Lung Fibroblasts: Emerging Roles of Cathepsin B and Cystatin, C. J. Biol. Chem. 2014, 289, 16239–16251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, C.; Rahim, S.; Arnold, J.; Hielscher, A. Loss of caveolin-1 alters extracellular matrix protein expression and ductal architecture in murine mammary glands. PLoS ONE 2017, 12, e0172067. [Google Scholar] [CrossRef] [Green Version]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Yasuoka, H.; Stolz, D.B.; Feghali-Bostwick, C.A. Decreased caveolin-1 levels contribute to fibrosis and deposition of extracellular IGFBP-5. J. Cell. Mol. Med. 2011, 15, 957–969. [Google Scholar] [CrossRef]
- Aga, M.; Bradley, J.M.; Wanchu, R.; Yang, Y.-F.; Acott, T.S.; Keller, K.E. Differential effects of caveolin-1 and -2 knockdown on aqueous outflow and altered extracellular matrix turnover in caveolin-silenced trabecular meshwork cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5497–5509. [Google Scholar] [CrossRef]
- Porter, K.; Hirt, J.; Stamer, W.D.; Liton, P.B. Autophagic dysregulation in glaucomatous trabecular meshwork cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Tholen, S.; Biniossek, M.L.; Gansz, M.; Ahrens, T.D.; Schlimpert, M.; Kizhakkedathu, J.N.; Reinheckel, T.; Schilling, O. Double deficiency of cathepsins B and L results in massive secretome alterations and suggests a degradative cathepsin-MMP axis. Cell. Mol. Life Sci. 2014, 71, 899–916. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Company | Catalog | Dilution |
|---|---|---|---|
| CTSB | Cell Signaling | 31718S | 1:1000 |
| Cav1 | Sigma | C3237 | 1:1000 |
| Annexin | Santa Cruz Biotechnology | Sc-28385 | 1:1000 |
| uPA | R&D | MAB9185 | 1:1000 |
| uPAR | R&D | MAB807 | 1:1000 |
| PAI-1 | Santa Cruz Biotechnology | sc-5297 | 1:250 |
| GAPDH | Santa Cruz Biotechnology | sc-47724 | 1:500 |
| CSTC | Millipore | ABC20 | 1:1000 |
| SMA | Sigma | A2547 | 1:1000 |
| FN1 | Santa Cruz Biotechnology | 31718S | 1:2000 |
| ACTB | Santa Cruz Biotechnology | Sc-69879 | 1:500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nettesheim, A.; Shim, M.S.; Dixon, A.; Raychaudhuri, U.; Gong, H.; Liton, P.B. Cathepsin B Localizes in the Caveolae and Participates in the Proteolytic Cascade in Trabecular Meshwork Cells. Potential New Drug Target for the Treatment of Glaucoma. J. Clin. Med. 2021, 10, 78. https://doi.org/10.3390/jcm10010078
Nettesheim A, Shim MS, Dixon A, Raychaudhuri U, Gong H, Liton PB. Cathepsin B Localizes in the Caveolae and Participates in the Proteolytic Cascade in Trabecular Meshwork Cells. Potential New Drug Target for the Treatment of Glaucoma. Journal of Clinical Medicine. 2021; 10(1):78. https://doi.org/10.3390/jcm10010078
Chicago/Turabian StyleNettesheim, April, Myoung Sup Shim, Angela Dixon, Urmimala Raychaudhuri, Haiyan Gong, and Paloma B. Liton. 2021. "Cathepsin B Localizes in the Caveolae and Participates in the Proteolytic Cascade in Trabecular Meshwork Cells. Potential New Drug Target for the Treatment of Glaucoma" Journal of Clinical Medicine 10, no. 1: 78. https://doi.org/10.3390/jcm10010078