
Sepsis as a Pan-Endocrine Illness—Endocrine Disorders in Septic Patients


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
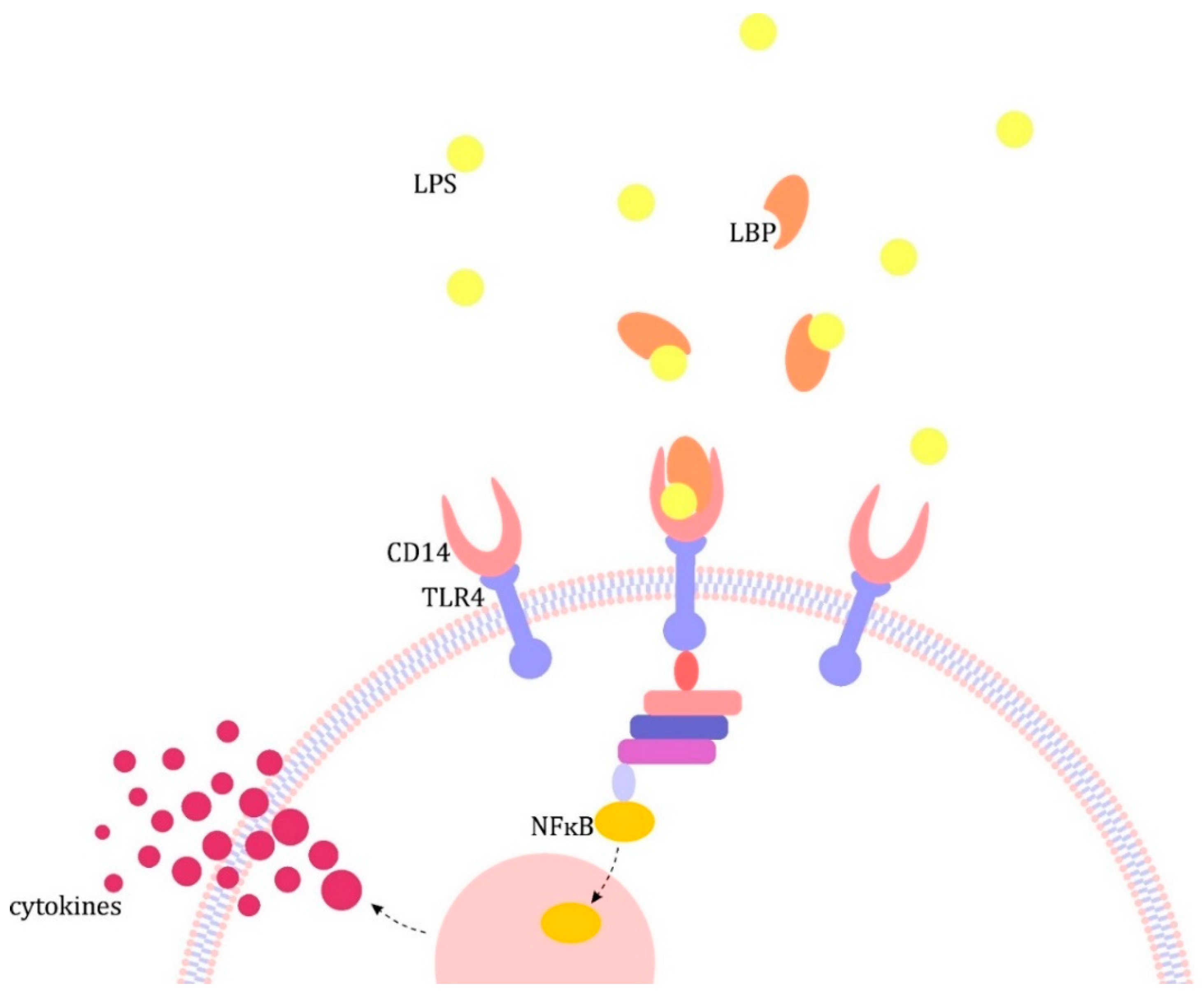
2. The Pathogenesis of Sepsis
3. Endocrine Disorders in the Course of Sepsis
3.1. Hypothalamic–Pituitary–Adrenal (HPA) Axis
3.2. Hypothalamic–Pituitary–Thyroid (HPT) Axis
3.3. Somatotropic Axis
3.4. Hypothalamic–Pituitary–Gonadal (HPG) Axis
3.5. Catecholamines, β-Adrenergic Stimulation and Insulin Resistance
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Carré, J.; Singer, M.; Moncada, S. Nitric Oxide. In Mechanisms of Sepsis-Induced Organ Dysfunction and Recovery; Springer: Berlin/Heidelberg, Germany, 2007; pp. 77–95. [Google Scholar]
- Singer, M. Dysfunction of the Bioenergetic Pathway; Springer: Berlin/Heidelberg, Germany, 2007; pp. 299–310. [Google Scholar]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Carré, J.E.; Singer, M. Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Et Biophys. Acta Bioenergy 2008, 1777, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kübler, A. Patogeneza. In Sepsa; Kübler, A., Ed.; Edra Urban & Partner: Wrocław, Poland, 2017; pp. 43–56. [Google Scholar]
- Bernard, C. An Introduction to the Study of Experimental Medicine; Dover Publications: New York, NY, USA, 1957. [Google Scholar]
- Cannon, W.B. Physiological regulation of normal states: Some tentative postulates concerning biological homeostatics. In A Charles Richet: Ses Amis, Ses Collègues, Ses Élèves; Pettit, A., Ed.; Les Éditions Médicales: Paris, France, 1926; p. 91. [Google Scholar]
- Cannon, W.B. Bodily Changes in Pain, Hunger, Fear and Rage, An Account of Recent Researches into the Function of Emotional Excitement; D Appleton & Company: New York, NY, USA, 1915. [Google Scholar]
- Goldstein, D.S.; Kopin, I.J. Evolution of concepts of stress. Stress 2007, 10, 109–120. [Google Scholar] [CrossRef]
- Gheorghiţă, V.; Barbu, A.E.; Gheorghiu, M.L.; Căruntu, F.A. Endocrine dysfunction in sepsis: A beneficial or deleterious host response? GERMS 2015, 5, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.S.; Stewart, P.M. Corticosteroid insufficiency in acutely ill patients. N. Engl. J. Med. 2003, 348, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, A.V.; Rivier, C.L. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: Actions and mechanisms of action. Physiol. Rev. 1999, 79, 1–71. [Google Scholar] [CrossRef] [Green Version]
- Annane, D.; Pastores, S.M.; Arlt, W.; Balk, R.A.; Beishuizen, A.; Briegel, J.; Carcillo, J.; Christ-Crain, M.; Cooper, M.S.; Marik, P.E.; et al. Critical illness-related corticosteroid insufficiency (CIRCI): A narrative review from a Multispecialty Task Force of the Society of Critical Care Medicine (SCCM) and the European Society of Intensive Care Medicine (ESICM). Intensive Care Med. 2017, 43, 1781–1792. [Google Scholar] [CrossRef]
- Kanczkowski, W.; Sue, M.; Zacharowski, K.; Reincke, M.; Bornstein, S.R. The role of adrenal gland microenvironment in the HPA axis function and dysfunction during sepsis. Mol. Cell. Endocrinol. 2015, 408, 241–248. [Google Scholar] [CrossRef]
- Mikhaylova, I.V.; Kuulasmaa, T.; Jääskeläinen, J.; Voutilainen, R. Tumor necrosis factor-α regulates steroidogenesis, apoptosis, and cell viability in the human adrenocortical cell line NCI-H295R. Endocrinology 2007, 148, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Engström, L.; Rosén, K.; Angel, A.; Fyrberg, A.; Mackerlova, L.; Konsman, J.P.; Engblom, D.; Blomqvist, A. Systemic immune challenge activates an intrinsically regulated local inflammatory circuit in the adrenal gland. Endocrinology 2008, 149, 1436–1450. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Singh, A.; Kral, T.; Solomon, S. The immune-hypothalamic-pituitary-adrenal axis. Endocr. Rev. 1989, 10, 92–112. [Google Scholar] [CrossRef]
- Ingels, C.; Gunst, J.; van den Berghe, G. Endocrine and Metabolic Alterations in Sepsis and Implications for Treatment. Crit. Care Clin. 2018, 34, 81–96. [Google Scholar] [CrossRef]
- Boonen, E.; Vervenne, H.; Meersseman, P.; Andrew, R.; Mortier, L.; Declercq, P.E.; Vanwijngaerden, Y.M.; Spriet, I.; Wouters, P.J.; Vander Perre, S.; et al. Reduced Cortisol Metabolism during Critical Illness. N. Engl. J. Med. 2013, 368, 1477–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonen, E.; Meersseman, P.; Vervenne, H.; Meyfroidt, G.; Guïza, F.; Wouters, P.J.; Veldhuis, J.D.; Van den Berghe, G. Reduced nocturnal ACTH-driven cortisol secretion during critical illness. Am. J. Physiol. Endocrinol. Metab. 2014, 306, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, D.; Vogt, B.; Escher, G.; Dick, B.; Reichen, J.; Frey, B.M.; Frey, F.J. Inhibition of 11β-hydroxysteroid dehydrogenase by bile acids in rats with cirrhosis. Hepatology 1999, 30, 623–629. [Google Scholar] [CrossRef]
- McNeilly, A.D.; Macfarlane, D.P.; O’Flaherty, E.; Livingstone, D.E.; Mitić, T.; McConnell, K.M.; McKenzie, S.M.; Davies, E.; Reynolds, R.M.; Thiesson, H.C.; et al. Bile acids modulate glucocorticoid metabolism and the hypothalamic-pituitary-adrenal axis in obstructive jaundice. J. Hepatol. 2010, 52, 705–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stauffer, A.T.; Rochat, M.K.; Dick, B.; Frey, F.J. Odermatt, Chenodeoxycholic acid and deoxycholic acid inhibit 11β-hydroxysteroid dehydrogenase type 2 and cause cortisol-induced transcriptional activation of the mineralocorticoid receptor. J. Biol. Chem. 2002, 277, 26286–26292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanwijngaerden, Y.M.; Wauters, J.; Langouche, L.; Vander Perre, S.; Liddle, C.; Coulter, S.; Vanderborght, S.; Roskams, T.; Wilmer, A.; Van den Berghe, G.; et al. Critical illness evokes elevated circulating bile acids related to altered hepatic transporter and nuclear receptor expression. Hepatology 2011, 54, 1741–1752. [Google Scholar] [CrossRef]
- Thomas, H. Sepsis: Bile acids promote inflammation in cholestasis-associated sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 324–325. [Google Scholar] [CrossRef] [PubMed]
- Pugeat, M.; Bonneton, A.; Perrot, D.; Rocle-Nicolas, B.; Lejeune, H.; Grenot, C.; Dechaud, H.; Brebant, C.; Motin, J.; Cuilleron, C.Y. Decreased immunoreactivity and binding activity of corticosteroid-binding globulin in serum in septic shock. Clin. Chem. 1989, 35, 1675–1679. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.J.; Kratzsch, J. Corticosteroid-binding globulin: Modulating mechanisms of bioavailability of cortisol and its clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Nenke, M.A.; Rankin, W.; Chapman, M.J.; Stevens, N.E.; Diener, K.R.; Hayball, J.D.; Lewis, J.G.; Torpy, D.J. Depletion of high-affinity corticosteroid-binding globulin corresponds to illness severity in sepsis and septic shock; Clinical implications. Clin. Endocrinol. 2015, 82, 801–807. [Google Scholar] [CrossRef]
- Ho, J.T.; Al-Musalhi, H.; Chapman, M.J.; Quach, T.; Thomas, P.D.; Bagley, C.J.; Lewis, J.G.; Torpy, D.J. Septic shock and sepsis: A comparison of total and free plasma cortisol levels. J. Clin. Endocrinol. Metab. 2006, 91, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molijn, G.J.; Spek, J.J.; Van Uffelen, J.C.; De Jong, F.H.; Brinkmann, A.O.; Bruining, H.A.; Lamberts, S.W.; Koper, J.W. Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. J. Clin. Endocrinol. Metab. 1995, 80, 1799–1803. [Google Scholar] [CrossRef]
- Cohen, J.; Pretorius, C.J.; Ungerer, J.P.; Cardinal, J.; Blumenthal, A.; Presneill, J.; Gatica-Andrades, M.; Jarrett, P.; Lassig-Smith, M.; Stuart, J.; et al. Glucocorticoid sensitivity is highly variable in critically ill patients with septic shock and is associated with disease severity. Crit. Care Med. 2016, 44, 1034–1041. [Google Scholar] [CrossRef]
- Alder, M.N.; Opoka, A.M.; Wong, H.R. The glucocorticoid receptor and cortisol levels in pediatric septic shock. Crit. Care 2018, 22, 244. [Google Scholar] [CrossRef] [Green Version]
- Jenniskens, M.; Weckx, R.; Dufour, T.; Vander Perre, S.; Pauwels, L.; Derde, S.; Téblick, A.; Güiza, F.; Van den Berghe, G.; Langouche, L. The hepatic glucocorticoid receptor is crucial for cortisol homeostasis and sepsis survival in humans and Male mice. Endocrinology 2018, 159, 2790–2802. [Google Scholar] [CrossRef]
- Abraham, M.N.; Jimenez, D.M.; Fernandes, T.D.; Deutschman, C.S. Cecal Ligation and Puncture Alters Glucocorticoid Receptor Expression. Crit. Care Med. 2018, 46, 797–804. [Google Scholar] [CrossRef]
- Ganesh, K.; Sharma, R.; Varghese, J.; Pillai, M.G.K. A profile of metabolic acidosis in patients with sepsis in an Intensive Care Unit setting. Int. J. Crit. Illn. Inj. Sci. 2016, 6, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardas, K.; Ilia, S.; Sertedaki, A.; Charmandari, E.; Briassouli, E.; Goukos, D.; Apostolou, K.; Psarra, K.; Botoula, E.; Tsagarakis, S.; et al. Increased glucocorticoid receptor expression in sepsis is related to heat shock proteins, cytokines, and cortisol and is associated with increased mortality. Intensive Care Med. Exp. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dendoncker, K.; Libert, C. Glucocorticoid resistance as a major drive in sepsis pathology. Cytokine Growth Factor Rev. 2017, 35, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Arafah, B.M. Review: Hypothalamic pituitary adrenal function during critical illness: Limitations of current assessment methods. J. Clin. Endocrinol. Metab. 2006, 91, 3725–3745. [Google Scholar] [CrossRef] [Green Version]
- Marik, P.E.; Pastores, S.M.; Annane, D.; Meduri, G.U.; Sprung, C.L.; Arlt, W.; Keh, D.; Briegel, J.; Beishuizen, A.; Dimopoulou, I.; et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit. Care Med. 2008, 36, 1937–1949. [Google Scholar] [CrossRef]
- Annane, D.; Pastores, S.M.; Rochwerg, B.; Arlt, W.; Balk, R.A.; Beishuizen, A.; Briegel, J.; Carcillo, J.; Christ-Crain, M.; Cooper, M.S.; et al. Guidelines for the diagnosis and management of Critical Illness-Related Corticosteroid Insufficiency (CIRCI) in critically ill patients (Part I): Society of Critical Care Medicine (SCCM) and European Society of Intensive Care Medicine (ESICM) 2017. Crit. Care Med. 2017, 45, 2078–2088. [Google Scholar] [CrossRef]
- Waterhouse, R. A case of suprarenal apoplexy. Lancet 1911, 177, 577–578. [Google Scholar] [CrossRef]
- Friderichsen, C. Nebennierenapoplexie bei kleinen Kindern. Jahrb. Kinderheilkd. 1918, 87, 109–125. [Google Scholar]
- Sharshar, T.; Annane, D.; de la Grandmaison, G.L.; Brouland, J.P.; Hopkinson, N.S.; Gray, F. The Neuropathology of Septic Shock. Brain Pathol. 2004, 14, 21–33. [Google Scholar] [CrossRef]
- McCann, S.M.; Kimura, M.; Karanth, S.; Yu, W.H.; Mastronardi, C.A.; Rettori, V. The Mechanism of Action of Cytokines to Control the Release of Hypothalamic and Pituitary Hormones in Infection. Ann. N. Y. Acad. Sci. 2006, 917, 4–18. [Google Scholar] [CrossRef]
- Polito, A.; Sonneville, R.; Guidoux, C.; Barrett, L.; Viltart, O.; Mattot, V.; Siami, S.; de la Grandmaison, G.L.; Chrétien, F.; Singer, M.; et al. Changes in CRH and ACTH Synthesis during Experimental and Human Septic Shock. PLoS ONE 2011, 6, e25905. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R. Predisposing Factors for Adrenal Insufficiency. N. Engl. J. Med. 2009, 360, 2328–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonen, E.; van den Berghe, G. Endocrine responses to critical illness: Novel insights and therapeutic implications. J. Clin. Endocrinol. Metab. 2014, 99, 1569–1582. [Google Scholar] [CrossRef] [Green Version]
- Polito, A.; de la Grandmaison, G.L.; Mansart, A.; Louiset, E.; Lefebvre, H.; Sharshar, T.; Annane, D. Human and experimental septic shock are characterized by depletion of lipid droplets in the adrenals. Intensive Care Med. 2010, 36, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Yen, P.M. Physiological and molecular basis of Thyroid hormone action. Physiol. Rev. 2001, 81, 1097–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalaki, M.; Vagenakis, A.G.; Makri, M.; Kalfarentzos, F.; Kyriazopoulou, V. Dissociation of the Early Decline in Serum T 3 Concentration and Serum IL-6 Rise and TNFα in Nonthyroidal Illness Syndrome Induced by Abdominal Surgery. J. Clin. Endocrinol. Metab. 2001, 86, 4198–4205. [Google Scholar] [CrossRef]
- Peeters, R.P.; Wouters, P.J.; Kaptein, E.; van Toor, H.; Visser, T.J.; van den Berghe, G. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J. Clin. Endocrinol. Metab. 2003, 88, 3202–3211. [Google Scholar] [CrossRef] [Green Version]
- Yildizdaş, D.; Onenli-Mungan, N.; Yapicioğlu, H.; Topaloğlu, A.K.; Sertdemir, Y.; Yüksel, B. Thyroid hormone levels and their relationship to survival in children with bacterial sepsis and septic shock. J. Pediatr. Endocrinol. Metab. 2004, 17, 1435–1442. [Google Scholar] [CrossRef]
- Rodriguez-Perez, A.; Palos-Paz, F.; Kaptein, E.; Visser, T.J.; Dominguez-Gerpe, L.; Alvarez-Escudero, J.; Lado-Abeal, J. Identification of molecular mechanisms related to nonthyroidal illness syndrome in skeletal muscle and adipose tissue from patients with septic shock. Clin. Endocrinol. 2008, 68, 821–827. [Google Scholar] [CrossRef]
- Yanni, G.N.; Destariani, C.P.; Lubis, A.N.; Deliana, M. Thyroid hormone profile in children with sepsis: Does euthyroid sick syndrome exist? Open Access Maced. J. Med. Sci. 2019, 7, 1110–1113. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Shi, S.; Shi, S. Sepsis leads to thyroid impairment and dysfunction in rat model. Tissue Cell 2016, 48, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Farwell, A.P. Euthyroid Sick Syndrome. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 1071–1080. [Google Scholar]
- Selvaraj, N.; Bobby, Z.; Sridhar, M.G. Is euthyroid sick syndrome a defensive mechanism against oxidative stress? Med. Hypotheses 2008, 71, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.L.; He, Q.; Hu, L.; He, C.Y.; Gao, L.C.; Young, C.A.; Chen, J.; Jiang, C.F.; Luo, X.F.; Zhou, Y.; et al. The relationship between inflammatory factors, oxidative stress and DIO-1 concentration in patients with chronic renal failure accompanied with or without euthyroid sick syndrome. J. Int. Med. Res. 2018, 46, 4061–4070. [Google Scholar] [CrossRef] [PubMed]
- van den Berghe, G. On the neuroendocrinopathy of critical illness: Perspectives for feeding and novel treatments. Am. J. Respir. Crit. Care Med. 2016, 194, 1337–1348. [Google Scholar] [CrossRef]
- Langouche, L.; Vander Perre, S.; Marques, M.; Boelen, A.; Wouters, P.J.; Casaer, M.P.; Van den Berghe, G. Impact of early nutrient restriction during critical illness on the nonthyroidal illness syndrome and its relation with outcome: A randomized, controlled clinical study. J. Clin. Endocrinol. Metab. 2013, 98, 1006–1013. [Google Scholar] [CrossRef] [Green Version]
- Mebis, L.; Eerdekens, A.; Güiza, F.; Princen, L.; Derde, S.; Vanwijngaerden, Y.M.; Vanhorebeek, I.; Darras, V.M.; Van den Berghe, G.; Langouche, L. Contribution of nutritional deficit to the pathogenesis of the nonthyroidal illness syndrome in critical illness: A rabbit model study. Endocrinology 2012, 153, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.F.; Docter, R.O.; Visser, T.J.; Krenning, E.P.; Bernard, B.; Van Toor, H.; De Jong, M.; Hennemann, G. Inhibition of thyroxine transport into cultured rat hepatocytes by serum of nonuremic critically ill patients: Effects of bilirubin and nonesterified fatty acids. J. Clin. Endocrinol. Metab. 1993, 76, 1165–1172. [Google Scholar] [CrossRef]
- den Brinker, M.; Joosten, K.F.; Visser, T.J.; Hop, W.C.; de Rijke, Y.B.; Hazelzet, J.A.; Boonstra, V.H.; Hokken-Koelega, A.C. Euthyroid sick syndrome in meningococcal sepsis: The impact of peripheral thyroid hormone metabolism and binding proteins. J. Clin. Endocrinol. Metab. 2005, 90, 5613–5620. [Google Scholar] [CrossRef] [Green Version]
- Van der Poll, T.; Van Zee, K.J.; Endert, E.; Coyle, S.M.; Stiles, D.M.; Pribble, J.P.; Catalano, M.A.; Moldawer, L.L.; Lowry, S.F. Interleukin-1 receptor blockade does not affect endotoxin-induced changes in plasma thyroid hormone and thyrotropin concentrations in man. J. Clin. Endocrinol. Metab. 1995, 80, 1341–1346. [Google Scholar] [CrossRef]
- Fliers, E.; Guldenaar, S.E.F.; Wiersinga, W.M.; Swaab, D.F. Decreased Hypothalamic Thyrotropin-Releasing Hormone Gene Expression in Patients with Nonthyroidal Illness 1. J. Clin. Endocrinol. Metab. 1997, 82, 4032–4036. [Google Scholar] [CrossRef]
- Van den Berghe, G.; De Zegher, F.; Veldhuis, J.D.; Wouters, P.; Gouwy, S.; Stockman, W.; Weekers, F.; Schetz, M.; Lauwers, P.; Bouillon, R.; et al. Thyrotrophin and prolactin release in prolonged critical illness: Dynamics of spontaneous secretion and effects of growth hormone-secretagogues. Clin. Endocrinol. 1997, 47, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Peeters, R.P.; Debaveye, Y.; Fliers, E.; Visser, T.J. Changes within the thyroid axis during critical illness. Crit. Care Clin. 2006, 22, 41–55. [Google Scholar] [CrossRef] [PubMed]
- van den Berghe, G. Non-thyroidal illness in the ICU: A syndrome with different faces. Thyroid 2014, 24, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; De Zegher, F.; Baxter, R.C.; Veldhuis, J.D.; Wouters, P.; Schetz, M.; Verwaest, C.; Van der Vorst, E.; Lauwers, P.; Bouillon, R.; et al. Neuroendocrinology of Prolonged Critical Illness: Effects of Exogenous Thyrotropin-Releasing Hormone and Its Combination with Growth Hormone Secretagogues 1. J. Clin. Endocrinol. Metab. 1998, 83, 309–319. [Google Scholar] [CrossRef]
- Angelousi, A.G.; Karageorgopoulos, D.E.; Kapaskelis, A.M.; Falagas, M.E. Association between thyroid function tests at baseline and the outcome of patients with sepsis or septic shock: A systematic review. Eur. J. Endocrinol. 2011, 164, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Foks, M.; Dudek, A.; Polok, K.; Nowak-Kózka, I.; Fronczek, J.; Szczeklik, W. Thyroid hormones as potential prognostic factors in sepsis. Anaesthesiol. Intensive Ther. 2019, 51, 205–209. [Google Scholar] [CrossRef]
- Borkowski, J.; Siemiatkowski, A.; Wołczyński, S.; Czaban, S.L.; Jedynak, M. Assessment of the release of thyroid hormones in septic shock—prognostic significance. Pol. Merkur. Lekarski 2005, 18, 45–48. [Google Scholar] [PubMed]
- Kothiwale, V.A.; Patil, P.; Gaur, S. Correlation of Thyroid Hormone Profile with the Acute Physiology and Chronic Health Evaluation II Score as a Prognostic Marker in Patients with Sepsis in the Intensive Care Unit. J. Assoc. Physicians India 2018, 66, 59–62. [Google Scholar] [PubMed]
- Giustina, A.; Veldhuis, J.D. Pathophysiology of the Neuroregulation of Growth Hormone Secretion in Experimental Animals and the Human 1. Endocr. Rev. 1998, 19, 717–797. [Google Scholar] [CrossRef]
- Perchard, R.; Clayton, P.E. Ghrelin and growth. Dev. Biol. Gastrointest. Horm. 2017, 32, 74–86. [Google Scholar] [CrossRef] [Green Version]
- Arvat, E.; Maccario, M.; Di Vito, L.; Broglio, F.; Benso, A.; Gottero, C.; Papotti, M.; Muccioli, G.; Dieguez, C.; Casanueva, F.F.; et al. Endocrine activities of ghrelin, a natural growth hormone secretagogue (GHS), in humans: Comparison and interactions with hexarelin, a nonnatural peptidyl GHS, and GH-releasing hormone. J. Clin. Endocrinol. Metab. 2001, 86, 1169–1174. [Google Scholar] [CrossRef]
- Carreira, M.C.; Crujeiras, A.B.; Andrade, S.; Monteiro, M.P.; Casanueva, F.F. Ghrelin as a GH-releasing factor. Endocr. Dev. 2013, 25, 49–58. [Google Scholar] [CrossRef]
- Dahn, M.S.; Lange, M.P.; Jacobs, L.A. Insulinlike Growth Factor 1 Production Is Inhibited in Human Sepsis. Arch. Surg. 1988, 123, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Dahn, M.S.; Lange, M.P. Systemic and splanchnic metabolic response to exogenous human growth hormone. Surgery 1998, 123, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Baxter, R.C.; Hawker, F.H.; To, C.; Stewart, P.M.; Holman, S.R. Thirty-day monitoring of insulin-like growth factors and their binding proteins in intensive care unit patients. Growth Horm. IGF Res. 1998, 8, 455–463. [Google Scholar] [CrossRef]
- Yumet, G.; Shumate, M.L.; Bryant, P.; Lin, C.M.; Lang, C.H.; Cooney, R.N. Tumor necrosis factor mediates hepatic growth hormone resistance during sepsis. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E472–E481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong-Brown, L.Q.; Brown, C.R.; Cooney, R.N.; Frost, R.A.; Lang, C.H. Sepsis-induced muscle growth hormone resistance occurs independently of STAT5 phosphorylation. Am. J. Physiol. Endocrinol. Metab. 2003, 285, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Langouche, L.; van den Berghe, G. The Dynamic Neuroendocrine Response to Critical Illness. Endocrinol. Metab. Clin. N. Am. 2006, 35, 777–791. [Google Scholar] [CrossRef]
- Adams, J.M.; Otero-Corchon, V.; Hammond, G.L.; Veldhuis, J.D.; Qi, N.; Low, M.J. Somatostatin is essential for the sexual dimorphism of gh secretion, corticosteroid-binding globulin production, and corticosterone levels in mice. Endocrinology 2015, 156, 1052–1065. [Google Scholar] [CrossRef] [Green Version]
- Lang, C.H.; Pollard, V.; Fan, J.; Traber, L.D.; Traber, D.L.; Frost, R.A.; Gelato, M.C.; Prough, D.S. Acute alterations in growth hormone-insulin-like growth factor axis in humans injected with endotoxin. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1997, 273, 371–378. [Google Scholar] [CrossRef]
- Baričević, I.; Jones, D.R.; Dordević, B.; Malenković, V.; Nedić, O. Differential influence of open surgery and sepsis on the circulating insulin-like growth factors and their binding proteins as representative metabolic markers. Clin. Biochem. 2007, 40, 1122–1128. [Google Scholar] [CrossRef]
- de Groof, F.; Joosten, K.F.; Janssen, J.A.; De Kleijn, E.D.; Hazelzet, J.A.; Hop, W.C.; Uitterlinden, P.; van Doorn, J.; Hokken-Koelega, A.C. Acute stress response in children with meningococcal sepsis: Important differences in the growth hormone/insulin-like growth factor I axis between nonsurvivors and survivors. J. Clin. Endocrinol. Metab. 2002, 87, 3118–3124. [Google Scholar] [CrossRef] [PubMed]
- Önenli-Mungan, N.; Yildizdas, D.; Yapicioglu, H.; Topaloglu, A.K.; Yüksel, B.; Özer, G. Growth hormone and insulin-like growth factor 1 levels and their relation to survival in children with bacterial sepsis and septic shock. J. Paediatr. Child Health 2004, 40, 221–226. [Google Scholar] [CrossRef]
- Papastathi, C.; Mavrommatis, A.; Mentzelopoulos, S.; Konstandelou, E.; Alevizaki, M.; Zakynthinos, S. Insulin-like Growth Factor I and its binding protein 3 in sepsis. Growth Horm. IGF Res. 2013, 23, 98–104. [Google Scholar] [CrossRef] [PubMed]
- van den Berghe, G. Dynamic neuroendocrine responses to critical illness. Front. Neuroendocrinol. 2002, 23, 370–391. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, C.; Cohen, A. Effect of IGF-1 on the balance between autophagy of dysfunctional mitochondria and apoptosis. FEBS Lett. 2004, 577, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shelat, H.; Geng, Y.J. IGF-1 prevents oxidative stress induced-apoptosis in induced pluripotent stem cells which is mediated by microRNA-1. Biochem. Biophys. Res. Commun. 2012, 426, 615–619. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, W.; Sun, R.; Liu, J.; Hong, J.; Li, Q.; Hu, B.; Gong, F. IGF‑1 may predict the severity and outcome of patients with sepsis and be associated with microRNA‑1 level changes. Exp. Ther. Med. 2017, 14, 797–804. [Google Scholar] [CrossRef] [Green Version]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Chang, L.; Zhao, J.; Yang, J.; Zhang, Z.; Du, J.; Tang, C. Therapeutic effects of ghrelin on endotoxic shock in rats. Eur. J. Pharmacol. 2003, 473, 171–176. [Google Scholar] [CrossRef]
- Maruna, P.; Gürlich, R.; Frasko, R.; Rosicka, M. Ghrelin and leptin elevation in postoperative intra-abdominal sepsis. Eur. Surg. Res. 2005, 37, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Waseem, T.; Duxbury, M.; Ito, H.; Ashley, S.W.; Robinson, M.K. Exogenous ghrelin modulates release of pro-inflammatory and anti-inflammatory cytokines in LPS-stimulated macrophages through distinct signaling pathways. Surgery 2008, 143, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Wang, P. Mechanism of the inhibitory effect of ghrelin in sepsis. Hepatic Med. Evid. Res. 2010, 2, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, S.X.; Shi, B.; Lou, X.L.; Liu, J.Q.; Ma, G.G.; Liang, D.Y.; Ma, S. Ghrelin protects small intestinal epithelium against sepsis-induced injury by enhancing the autophagy of intestinal epithelial cells. Biomed. Pharmacother. 2016, 83, 1315–1320. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, W.L.; Aziz, M.; Ma, G.; Wang, P. Therapeutic effect of human ghrelin and growth hormone: Attenuation of immunosuppression in septic aged rats. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2584–2593. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.I. Altered gonadal steroidogenesis in critical illness: Is treatment with anabolic steroids indicated? Best Pract. Res. Clin. Endocrinol. Metab. 2001, 15, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Samson, W.K.; Taylor, M.M.; Baker, J.R. Prolactin-releasing peptides. Regul. Pept. 2003, 114, 1–5. [Google Scholar] [CrossRef]
- Savino, W. Prolactin: An Immunomodulator in Health and Disease. Front. Horm. Res. 2017, 48, 69–75. [Google Scholar] [CrossRef]
- Guo, H.; Calkins, J.H.; Sigel, M.M.; Lin, T. Interleukin-2 is a potent inhibitor of Leydig cell steroidogenesis. Endocrinology 1990, 127, 1234–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Q.; Hawker, F.; McWilliam, D.; Bangah, M.; Burger, H.; Handelsman, D.J. Circulating immunoreactive inhibin and testosterone levels in men with critical illness. Clin. Endocrinol. 1992, 36, 399–404. [Google Scholar] [CrossRef]
- Wang, C.; Chan, V.; Yeung, R.T. Effect of surgical stress on pituitary-testicular function. Clin. Endocrinol. 1978, 9, 255–266. [Google Scholar] [CrossRef]
- Wang, C.; Chan, V.; Tse, T.F.; Yeung, R.T.T. Effect of acute myocardial infarction on pituitary-testicular function. Clin. Endocrinol. 1978, 9, 249–253. [Google Scholar] [CrossRef]
- Sharshar, T.; Bastuji-Garin, S.; De Jonghe, B.; Stevens, R.D.; Polito, A.; Maxime, V.; Rodriguez, P.; Cerf, C.; Outin, H.; Touraine, P.; et al. Hormonal status and ICU-acquired paresis in critically ill patients. Intensive Care Med. 2010, 36, 1318–1326. [Google Scholar] [CrossRef]
- Christeff, N.; Benassayag, C.; Carli-Vielle, C.; Carli, A.; Nunez, E.A. Elevated oestrogen and reduced testosterone levels in the serum of male septic shock patients. J. Steroid Biochem. 1988, 29, 435–440. [Google Scholar] [CrossRef]
- Woolf, P.D.; Hamill, R.W.; McDonald, J.V.; Lee, L.A.; Kelly, M. Transient hypogonadotropic hypogonadism caused by critical illness. J. Clin. Endocrinol. Metab. 1985, 60, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Vanhorebeek, I.; Langouche, L.; den van den Berghe, G. Endocrine aspects of acute and prolonged critical illness. Nat. Clin. Pract. Endocrinol. Metab. 2006, 2, 20–31. [Google Scholar] [CrossRef]
- Berghe, G.; Zegher, F.; Lauwers, P.; Veldhuls, J.D. Luteinizing hormone secretion and hypoandrogenaemia in critically ill men: Effect of dopamine. Clin. Endocrinol. 1994, 41, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; Weekers, F.; Baxter, R.C.; Wouters, P.; Iranmanesh, A.; Bouillon, R.; Veldhuis, J.D. Five-Day Pulsatile Gonadotropin-Releasing Hormone Administration Unveils Combined Hypothalamic-Pituitary-Gonadal Defects Underlying Profound Hypoandrogenism in Men with Prolonged Critical Illness 1. J. Clin. Endocrinol. Metab. 2001, 86, 3217–3226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechanick, J.I.; Nierman, D.M. Gonadal steroids in critical illness. Crit. Care Clin. 2006, 22, 87–103. [Google Scholar] [CrossRef]
- Breen, K.M.; Karsch, F.J. Does Cortisol Inhibit Pulsatile Luteinizing Hormone Secretion at the Hypothalamic or Pituitary Level? Endocrinology 2004, 145, 692–698. [Google Scholar] [CrossRef]
- Breen, K.M.; Stackpole, C.A.; Clarke, I.J.; Pytiak, A.V.; Tilbrook, A.J.; Wagenmaker, E.R.; Young, E.A.; Karsch, F.J. Does the type II glucocorticoid receptor mediate cortisol-induced suppression in pituitary responsiveness to gonadotropin-releasing hormone? Endocrinology 2004, 145, 2739–2746. [Google Scholar] [CrossRef] [Green Version]
- Kalantaridou, S.N.; Makrigiannakis, A.; E Zoumakis, G.P. Chrousos, Stress and the female reproductive system. J. Reprod. Immunol. 2004, 62, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Vardas, K.; Apostolou, K.; Briassouli, E.; Goukos, D.; Psarra, K.; Botoula, E.; Tsagarakis, S.; Magira, E.; Routsi, C.; Nanas, S.; et al. Early Response Roles for Prolactin Cortisol and Circulating and Cellular Levels of Heat Shock Proteins 72 and 90α in Severe Sepsis and SIRS. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Moss, M. Epidemiology of Sepsis: Race, Sex, and Chronic Alcohol Abuse. Clin. Infect. Dis. 2005, 41, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Sakr, Y.; Elia, C.; Mascia, L.; Barberis, B.; Cardellino, S.; Livigni, S.; Fiore, G.; Filippini, C.; Ranieri, V.M. The influence of gender on the epidemiology of and outcome from severe sepsis. Crit. Care 2013, 17, 50. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, C.; Thomas-Rueddel, D.O.; Hartmann, M.; Hartog, C.S.; Welte, T.; Heublein, S.; Dennler, U.; Reinhart, K. Fallzahlen und sterblichkeitsraten von sepsis-patienten im krankenhaus. Dtsch. Arztebl. Int. 2016, 113, 159–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, N.; Jamil, B.; Siddiqui, S.; Talat, N.; Khan, F.A.; Hussain, R. Mortality in sepsis and its relationship with gender. Pakistan, J. Med. Sci. 2015, 31, 1201–1206. [Google Scholar] [CrossRef]
- Xu, J.; Tong, L.; Yao, J.; Guo, Z.; Lui, K.Y.; Hu, X.; Cao, L.; Zhu, Y.; Huang, F.; Guan, X.; et al. Association of Sex with Clinical Outcome in Critically Ill Sepsis Patients: A Retrospective Analysis of the Large Clinical Database MIMIC-III. Shock 2019, 52, 146–151. [Google Scholar] [CrossRef]
- Sunden-Cullberg, J.; Nilsson, A.; Inghammar, M. Sex-based differences in ED management of critically ill patients with sepsis: A nationwide cohort study. Intensive Care Med. 2020, 46, 727–736. [Google Scholar] [CrossRef] [Green Version]
- de Farias, L.M.; de Brito, D.; Sales, J.P.; Rodrigues, R.S.; de Meneses, F.A. Gender and mortality in sepsis: Do sex hormones impact the outcome? Rev. Bras. Ter. Intensiva 2011, 23, 297–303. [Google Scholar]
- El-Lakany, M.A.; Fouda, M.A.; El-Gowelli, H.M.; El-Gowilly, S.M.; El-Mas, M.M. Gonadal hormone receptors underlie the resistance of female rats to inflammatory and cardiovascular complications of endotoxemia. Eur. J. Pharmacol. 2018, 823, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Saia, R.S.; Garcia, F.M.; Cárnio, E.C. Estradiol protects female rats against sepsis induced by Enterococcus faecalis improving leukocyte bactericidal activity. Steroids 2015, 102, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Surewaard, B.G.; Wong, C.H.; Guettler, C.; Petri, B.; Burkhard, R.; Wyss, M.; Le Moual, H.; Devinney, R.; Thompson, G.C.; et al. Sex-hormone-driven innate antibodies protect females and infants against EPEC infection. Nat. Immunol. 2018, 19, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Martínez, E.R.; García-Gómez, E.; Camacho-Arroyo, I.; González-Pedrajo, B. Sexual dimorphism in bacterial infections. Biol. Sex Differ. 2018, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Norbury, W.B.; Jeschke, M.G.; Herndon, D.N. Metabolism modulators in sepsis: Propranolol. Crit. Care Med. 2007, 35, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.H. Sepsis-induced insulin resistance in rats is mediated by a β-adrenergic mechanism. Am. J. Physiol. Endocrinol. Metab. 1992, 263, 703–711. [Google Scholar] [CrossRef]
- Deibert, D.C.; DeFronzo, R.A. Epinephrine-induced insulin resistance in man. J. Clin. Investig. 1980, 65, 717–721. [Google Scholar] [CrossRef]
- Andersen, S.K.; Gjedsted, J.; Christiansen, C.; Tønnesen, E. The roles of insulin and hyperglycemia in sepsis pathogenesis. J. Leukoc. Biol. 2004, 75, 413–421. [Google Scholar] [CrossRef]
- Lönnroth, P.; Smith, U. β-Adrenergic dependent downregulation of insulin binding in rat adipocytes. Biochem. Biophys. Res. Commun. 1983, 112, 972–979. [Google Scholar] [CrossRef]
- Chu, C.A.; Sindelar, D.K.; Igawa, K.; Sherck, S.; Neal, D.W.; Emshwiller, M.; Cherrington, A.D. The direct effects of catecholamines on hepatic glucose production occur via α1- and β2-receptors in the dog. Am. J. Physiol. Endocrinol. Metab. 2000, 279. [Google Scholar] [CrossRef] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 281, 785–789. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasyluk, W.; Wasyluk, M.; Zwolak, A. Sepsis as a Pan-Endocrine Illness—Endocrine Disorders in Septic Patients. J. Clin. Med. 2021, 10, 2075. https://doi.org/10.3390/jcm10102075
Wasyluk W, Wasyluk M, Zwolak A. Sepsis as a Pan-Endocrine Illness—Endocrine Disorders in Septic Patients. Journal of Clinical Medicine. 2021; 10(10):2075. https://doi.org/10.3390/jcm10102075
Chicago/Turabian StyleWasyluk, Weronika, Martyna Wasyluk, and Agnieszka Zwolak. 2021. "Sepsis as a Pan-Endocrine Illness—Endocrine Disorders in Septic Patients" Journal of Clinical Medicine 10, no. 10: 2075. https://doi.org/10.3390/jcm10102075