
Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications

 ,
,  , , ,
, , ,  ,
,  and
and
Abstract
1. Introduction
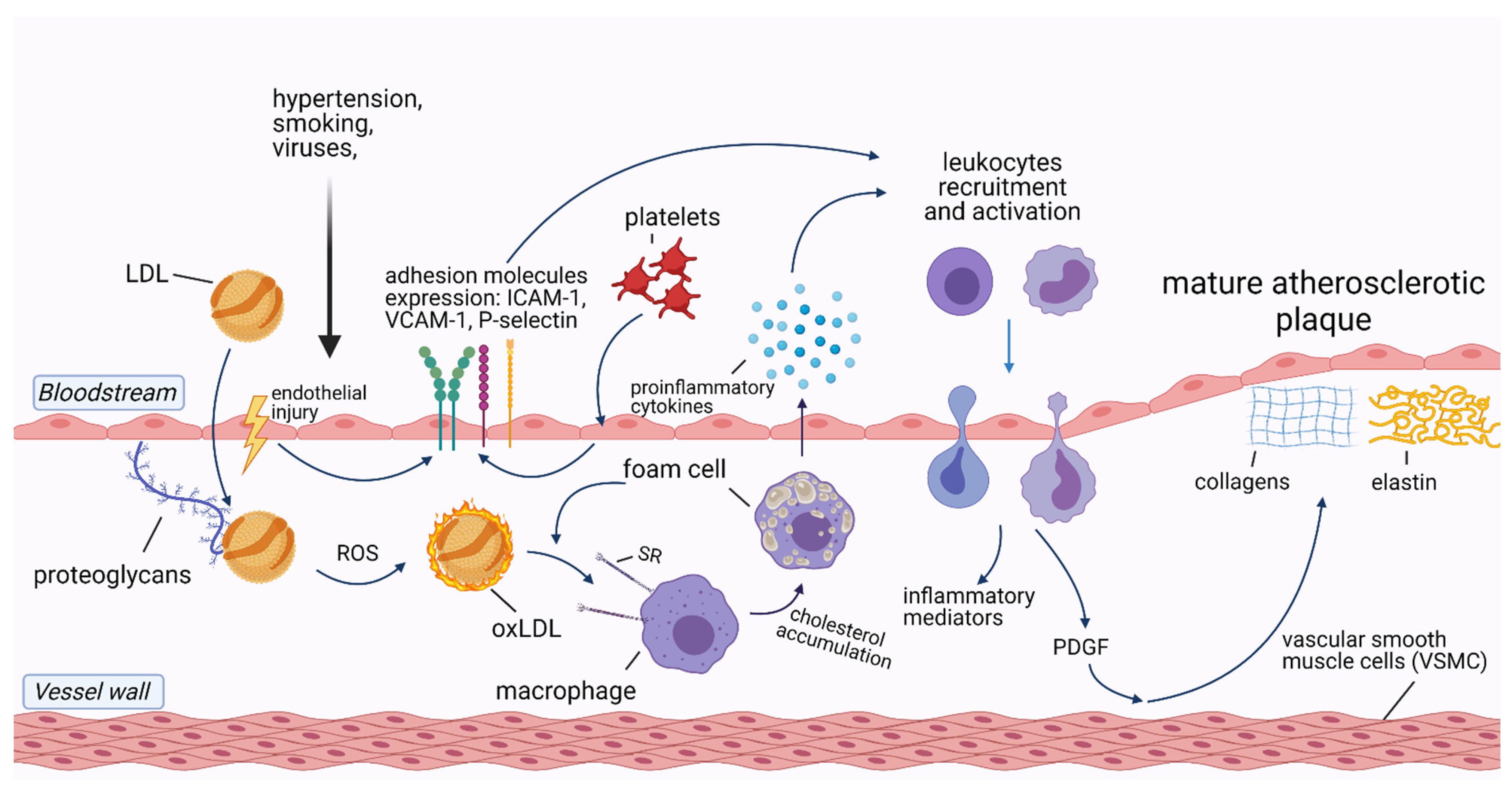
2. The Role of Inflammation in Cardiovascular Diseases
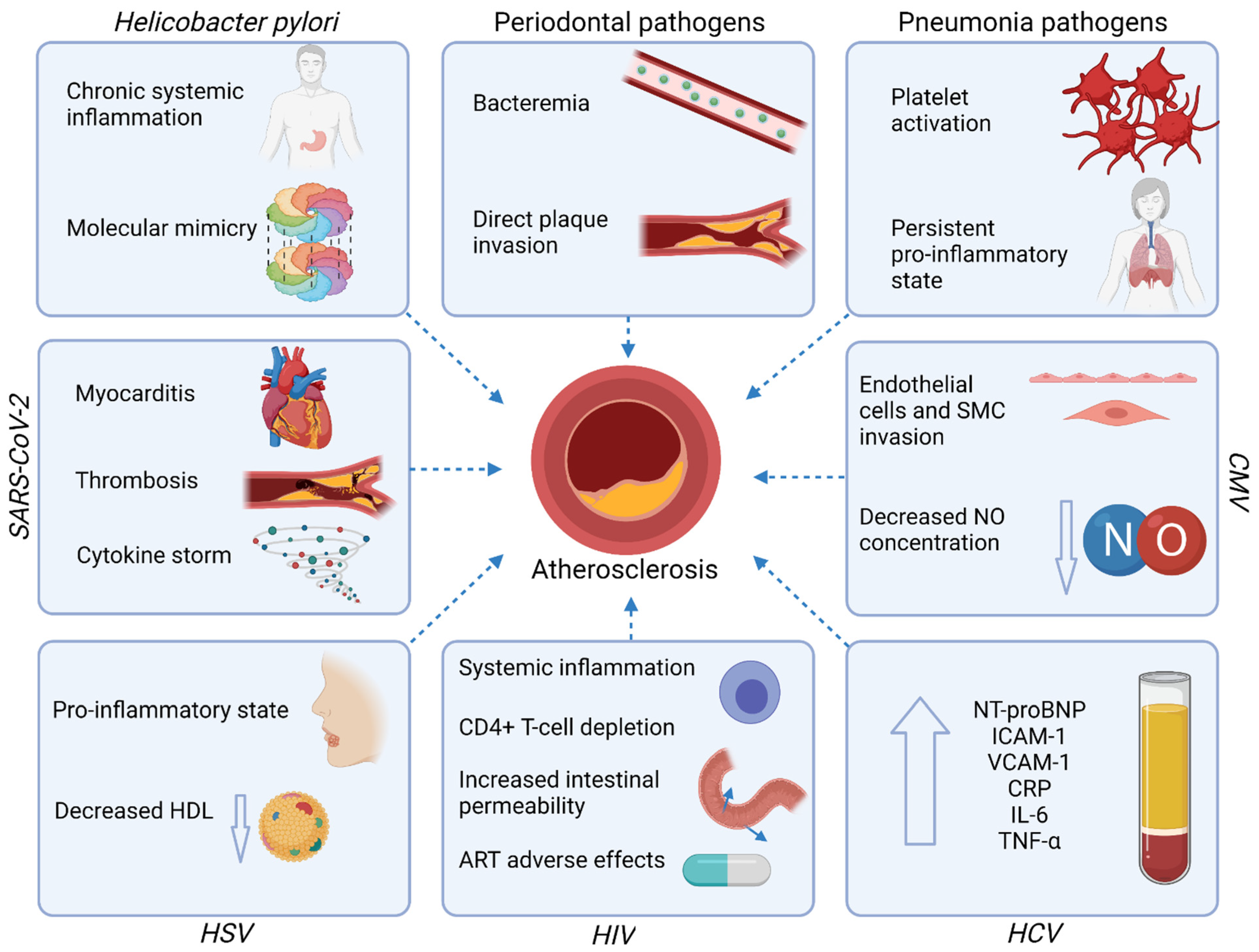
3. Infections and Atherosclerotic Cardiovascular Diseases
3.1. Gastrointestinal Tract Infections
3.1.1. Periodontal Disease
3.1.2. Helicobacter Pylori
3.1.3. Hepatitis C Virus (HCV)
3.2. Respiratory Tract Infections
3.2.1. Pneumonia
3.2.2. Cytomegalovirus (CMV)
3.2.3. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2)
3.3. Immune System Infections
Human Immunodeficiency Virus (HIV)
3.4. Dermatologic Infections
Herpes Simplex Virus (HSV)
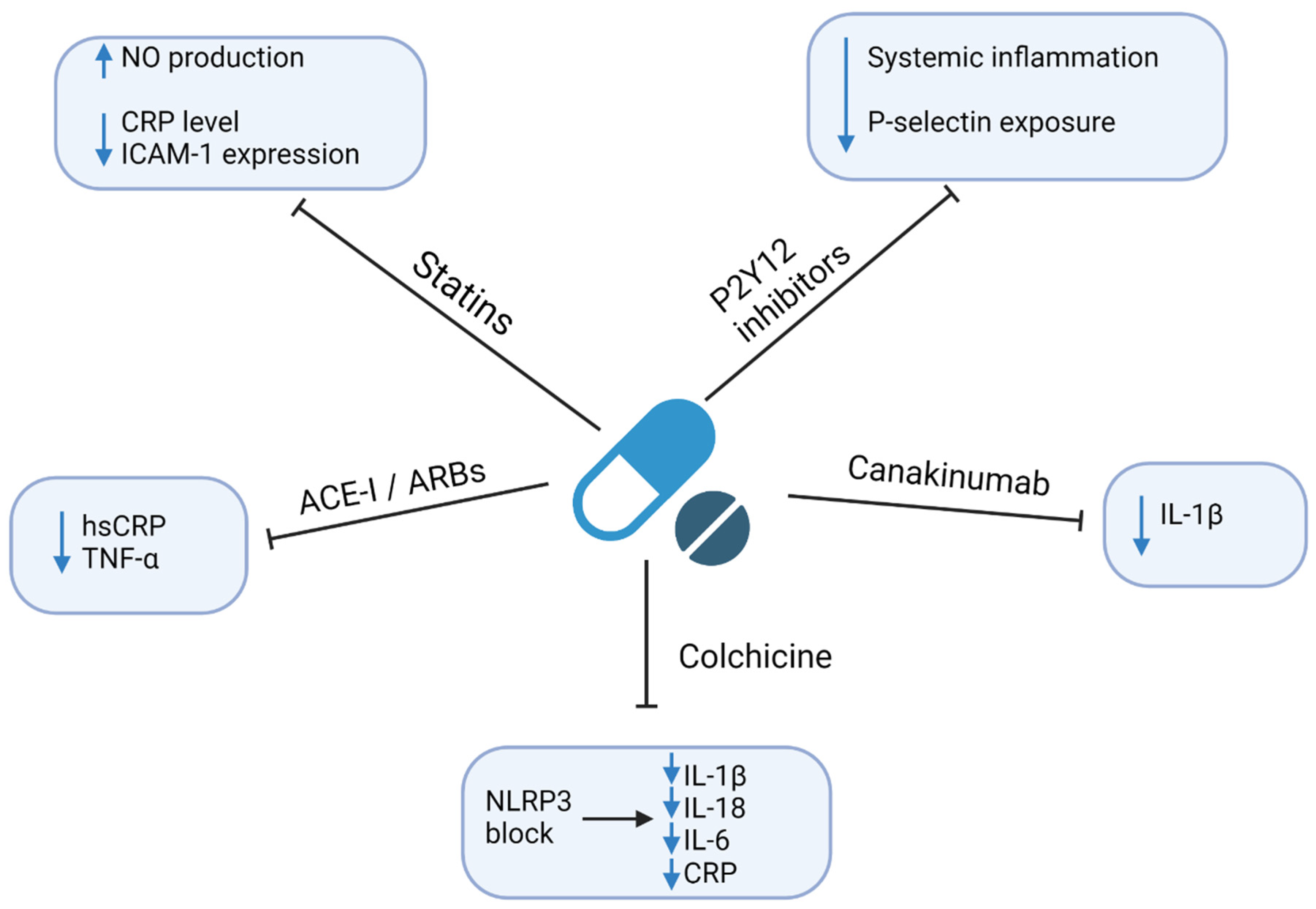
4. Therapeutic Implications
4.1. Statins
4.2. P2Y12 Inhibitors
4.3. Angiotensin-Converting Enzyme Inhibitors (ACE-I) and Angiotensin Receptor Blockers (ARBs)
4.4. Colchicine
4.5. Anti-Cytokine Drugs
4.6. Methotrexate
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Data. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 20 March 2021).
- Mihaylova, B.; Emberson, J.; Blackwell, L.; Keech, A.; Simes, J.; Barnes, E.H.; Voysey, M.; Gray, A.; Collins, R.; Baigent, C.; et al. The Effects of Lowering LDL Cholesterol with Statin Therapy in People at Low Risk of Vascular Disease: Meta-Analysis of Individual Data from 27 Randomised Trials. Lancet 2012, 380, 581–590. [Google Scholar] [CrossRef]
- Kaasenbrood, L.; Boekholdt, S.M.; Van Der Graaf, Y.; Ray, K.K.; Peters, R.J.G.; Kastelein, J.J.P.; Amarenco, P.; Larosa, J.C.; Cramer, M.J.M.; Westerink, J.; et al. Distribution of Estimated 10-Year Risk of Recurrent Vascular Events and Residual Risk in a Secondary Prevention Population. Circulation 2016, 134, 1419–1429. [Google Scholar] [CrossRef]
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Kränkel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle Factors and High-Risk Atherosclerosis: Pathways and Mechanisms beyond Traditional Risk Factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Corrado, E.; Novo, S. Role of Inflammation and Infection in Vascular Disease. Acta Chir. Belg. 2005, 105, 567–579. [Google Scholar] [CrossRef]
- Alfarisi, H.A.H.; Mohamed, Z.B.H.; Ibrahim, M. Bin. Basic Pathogenic Mechanisms of Atherosclerosis. Egypt. J. Basic Appl. Sci. 2020, 7, 116–125. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of Endothelial Dysfunction in Atherosclerosis. Circulation 2004, 109 (Suppl. 23). [Google Scholar] [CrossRef] [PubMed]
- Leishman, S.J.; Do, H.L.; Ford, P.J. Cardiovascular Disease and the Role of Oral Bacteria. J. Oral Microbiol. 2010, 2, 5781. [Google Scholar] [CrossRef]
- Xu, Z.; Li, J.; Wang, H.; Xu, G. Helicobacter Pylori Infection and Atherosclerosis: Is There a Causal Relationship? Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2293–2301. [Google Scholar] [CrossRef]
- Lebedeva, A.M.; Shpektor, A.V.; Vasilieva, E.Y.; Margolis, L.B. Cytomegalovirus Infection in Cardiovascular Diseases. Biochemistry 2018, 83, 1437–1447. [Google Scholar] [CrossRef]
- Restrepo, M.I.; Reyes, L.F. Pneumonia as a Cardiovascular Disease. Respirology 2018, 23, 250–259. [Google Scholar] [CrossRef]
- Berquist, V.; Hoy, J.F.; Trevillyan, J.M. Contribution of Common Infections to Cardiovascular Risk in HIV-Positive Individuals. AIDS Rev. 2017, 19, 72–80. [Google Scholar] [PubMed]
- Wu, Y.P.; Sun, D.D.; Wang, Y.; Liu, W.; Yang, J. Herpes Simplex Virus Type 1 and Type 2 Infection Increases Atherosclerosis Risk: Evidence Based on a Meta-Analysis. BioMed Res. Int. 2016, 2016, 2630865. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, B.P.; Sweitzer, N.K.; Indik, J.H.; Acharya, D.; William, P. SARS-CoV-2 Infection and Cardiovascular Disease: COVID-19 Heart. Heart Lung Circ. 2020, 29, 973–987. [Google Scholar] [CrossRef]
- WHO Data. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 2 May 2021).
- Williams, K.J.; Tabas, I. The Response-to-Retention Hypothesis of Early Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Hermansson, A. The Immune System in Atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M. Inflammation and Atherothrombosis. From Population Biology and Bench Research to Clinical Practice. J. Am. Coll. Cardiol. 2006, 48, 33–46. [Google Scholar] [CrossRef]
- Lorenzatti, A.J.; Servato, M.L. New Evidence on the Role of Inflammation in CVD Risk. Curr. Opin. Cardiol. 2019, 34, 418–423. [Google Scholar] [CrossRef]
- Raggi, P.; Genest, J.; Giles, J.T.; Rayner, K.J.; Dwivedi, G.; Beanlands, R.S.; Gupta, M. Role of Inflammation in the Pathogenesis of Atherosclerosis and Therapeutic Interventions. Atherosclerosis 2018, 276, 98–108. [Google Scholar] [CrossRef]
- Gisterå, A.; Robertson, A.K.L.; Andersson, J.; Ketelhuth, D.F.J.; Ovchinnikova, O.; Nilsson, S.K.; Lundberg, A.M.; Li, M.O.; Flavell, R.A.; Hansson, G.K. Transforming Growth Factor-β Signaling in T Cells Promotes Stabilization of Atherosclerotic Plaques through an Interleukin-17-Dependent Pathway. Sci. Transl. Med. 2013, 5, 18–23. [Google Scholar] [CrossRef]
- Libby, P.; Hansson, G.K. Taming Immune and Inflammatory Responses to Treat Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 173–176. [Google Scholar] [CrossRef]
- Lordan, R.; Tsoupras, A.; Zabetakis, I. Platelet Activation and Prothrombotic Mediators at the Nexus of Inflammation and Atherosclerosis: Potential Role of Antiplatelet Agents. Blood Rev. 2021, 45, 100694. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Rogula, S.; Szarpak, Ł.; Filipiak, K.J. LDL-Cholesterol and Platelets: Insights into Their Interactions in Atherosclerosis. Life 2021, 11, 39. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Künzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef] [PubMed]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, Functions, and Clinical Relevance of Extracellular Vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef]
- Hafiane, A.; Daskalopoulou, S.S. Extracellular Vesicles Characteristics and Emerging Roles in Atherosclerotic Cardiovascular Disease. Metabolism 2018, 85, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Pluta, K.; Solarska, K.; Rydz, B.; Eyileten, C.; Postula, M.; Van Der Pol, E.; Nieuwland, R.; Budnik, M.; Kochanowski, J.; et al. Plasma Concentrations of Extracellular Vesicles Are Decreased in Patients with Post-Infarct Cardiac Remodelling. Biology 2021, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Van Der Pol, E.; Nieuwland, R.; Stępień, E. Extracellular Vesicles in Post-Infarct Ventricular Remodelling. Kardiol. Pol. 2018, 76, 69–76. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Siljander, P.R.M. Platelet-Derived Extracellular Vesicles. In Platelets, 4th ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 401–416. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in Cardiovascular Biology and Disease. Adv. Clin. Exp. Med. 2017, 26, 865–874. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, J.; Zhang, F.; Lei, Q.; Zhang, T.; Li, K.; Guo, J.; Hong, Y.; Bu, G.; Lv, X.; et al. MicroRNA-181a-5p and MicroRNA-181a-3p Cooperatively Restrict Vascular Inflammation and Atherosclerosis. Cell Death Dis. 2019, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Soh, J. MicroRNA-30c Reduces Hyperlipidemia and Atherosclerosis by Decreasing Lipid Synthesis and Lipoprotein Secretion. Physiol. Behav. 2016, 176, 100–106. [Google Scholar] [CrossRef]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.A.; Heyll, K.; Noels, H.; Hristov, M.; et al. MicroRNA-126-5p Promotes Endothelial Proliferation and Limits Atherosclerosis by Suppressing Dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.C.; Tang, Y.Y.; Peng, J.; Zhao, G.J.; Yang, J.; Yao, F.; Ouyang, X.P.; He, P.P.; Xie, W.; Tan, Y.L.; et al. MicroRNA-19b Promotes Macrophage Cholesterol Accumulation and Aortic Atherosclerosis by Targeting ATP-Binding Cassette Transporter A1. Atherosclerosis 2014, 236, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. Microrna-33 regulates macrophage autophagy in atherosclerosis. Arterioscler. Thrombo. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Potteaux, S.; Vion, A.C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.L.; et al. Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and Atherosclerosis in Mice. Circ. Res. 2014, 114, 434–443. [Google Scholar] [CrossRef]
- Hu, Y.W.; Hu, Y.R.; Zhao, J.Y.; Li, S.F.; Ma, X.; Wu, S.G.; Lu, J.B.; Qiu, Y.R.; Sha, Y.H.; Wang, Y.C.; et al. An Agomir of MiR-144-3p Accelerates Plaque Formation through Impairing Reverse Cholesterol Transport and Promoting pro-Inflammatory Cytokine Production. PLoS ONE 2014, 9, e94997. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 Promotes Atherosclerosis by Repressing Bcl6 in Macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Nazari-Jahantigh, M.; Chan, L.; Zhu, M.; Heyll, K.; Corbalán-Campos, J.; Hartmann, P.; Thiemann, A.; Weber, C.; Schober, A. The MicroRNA-342-5p Fosters Inflammatory Macrophage Activation through an Akt1- and MicroRNA-155-Dependent Pathway during Atherosclerosis. Circulation 2013, 127, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Vacante, F.; Denby, L.; Sluimer, J.C.; Baker, A.H. The Function of MiR-143, MiR-145 and the MiR-143 Host Gene in Cardiovascular Development and Disease. Vascul. Pharmacol. 2019, 112, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Gozdowska, R.; Makowska, A.; Gąsecka, A.; Chabior, A.; Marchel, M. Circulating MicroRNA in Heart Failure—Practical Guidebook to Clinical Application. Cardiol Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tomaniak, M.; Sygitowicz, G.; Filipiak, K.J.; Błaszczyk, O.; Kołtowski, L.; Gasecka, A.; Kochanowski, J.; Sitkiewicz, D. Dysregulations of MiRNAs and Galectin-3 May Underlie Left Ventricular Dilatation in Patients with Systolic Heart Failure. Kardiol. Pol. 2018, 76, 1012–1014. [Google Scholar] [CrossRef] [PubMed]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef]
- Adawi, M.; Firas, S.; Blum, A. Rheumatoid Arthritis and Atherosclerosis. Isr. Med. Assoc. J. 2019, 21, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, D.; Cannavo, A.; Spagnuolo, G.; Ferrara, N.; Cittadini, A.; Rengo, C.; Rengo, G. Periodontal Disease: A Risk Factor for Diabetes and Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 1414. [Google Scholar] [CrossRef]
- Tomita, S.; Komiya-Ito, A.; Imamura, K.; Kita, D.; Ota, K.; Takayama, S.; Makino-Oi, A.; Kinumatsu, T.; Ota, M.; Saito, A. Prevalence of Aggregatibacter Actinomycetemcomitans, Porphyromonas Gingivalis and Tannerella Forsythia in Japanese Patients with Generalized Chronic and Aggressive Periodontitis. Microb. Pathog. 2013, 61, 11–15. [Google Scholar] [CrossRef]
- Larvin, H.; Kang, J.; Aggarwal, V.R.; Pavitt, S.; Wu, J. Risk of Incident Cardiovascular Disease in People with Periodontal Disease: A Systematic Review and Meta-Analysis. Clin. Exp. Dent. Res. 2021, 7, 109–122. [Google Scholar] [CrossRef]
- Carrizales-Sepúlveda, E.F.; Ordaz-Farías, A.; Vera-Pineda, R.; Flores-Ramírez, R. Periodontal Disease, Systemic Inflammation and the Risk of Cardiovascular Disease. Heart Lung Circ. 2018, 27, 1327–1334. [Google Scholar] [CrossRef]
- Sanz, M.; Marco del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases: Consensus Report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef]
- Schenkein, H.A.; Loos, B.G. Inflammatory Mechanisms Linking Periodontal Diseases to Cardiovascular Diseases. J. Clin. Periodontol. 2013, 40, S51–S69. [Google Scholar] [CrossRef] [PubMed]
- Roca-Millan, E.; González-Navarro, B.; Del Mar Sabater-Recolons, M.; Marí-Roig, A.; Jané-Salas, E.; López-López, J. Periodontal Treatment on Patients with Cardiovascular Disease: Systematic Review and Meta-Analysis. Med. Oral Patol. Oral Cir. Bucal 2018, 23, E681–E690. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.G.; Schmidt, M.M.; Lopes, R.D.; Dipp, T.; Feijó, I.P.; Schmidt, K.E.S.; Gazeta, C.A.; Azeredo, M.L.; Markoski, M.; Pellanda, L.C.; et al. Treating Periodontal Disease in Patients with Myocardial Infarction: A Randomized Clinical Trial. Eur. J. Intern. Med. 2020, 71, 76–80. [Google Scholar] [CrossRef]
- Tonetti, M.S.; D’Aiuto, F.; Nibali, L. Treatment of Periodontitis and Endothelial Function. Jpn. J. Chest Dis. 2008, 67, 353. [Google Scholar] [CrossRef]
- Sharma, S.; Sridhar, S.; Mcintosh, A.; Messow, C.-M.; Aguilera, E.M.; Del Pinto, R.; Pietropaoli, D.; Górska, R.; Siedlinski, M.; Maffia, P.; et al. Periodontal Therapy and Treatment of Hypertensio—Alternative to the Pharmacological Approach. A Systematic Review and Meta-Analysis. Pharmacol. Res. 2021, 19, 105511. [Google Scholar] [CrossRef]
- Czerniuk, M.R.; Górska, R.; Filipiak, K.J.; Opolski, G. C-Reactive Protein in Patients with Coexistent Periodontal Disease and Acute Coronary Syndromes. J. Clin. Periodontol. 2006, 33, 415–420. [Google Scholar] [CrossRef]
- Surma, S.; Romańczyk, M.; Witalińska-Łabuzek, J.; Czerniuk, M.R.; Łabuzek, K.; Filipiak, K.J. Periodontitis, Blood Pressure, and the Risk and Control of Arterial Hypertension: Epidemiological, Clinical, and Pathophysiological Aspects-Review of the Literature and Clinical Trials. Curr. Hypertens. Rep. 2021, 23, 27. [Google Scholar] [CrossRef]
- Cave, D.R.; Go, M.; Cutler, A.; Goldstein, J.; Dunn, B.; Mobley, H.; Barkin, J.; Fennerty, B.; Hunt, R.; Peura, D.; et al. Transmission and Epidemiology of Helicobacter Pylori. Am. J. Med. 1996, 100, S12–S18. [Google Scholar] [CrossRef]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter Pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef]
- Fang, Y.; Fan, C.; Xie, H.; Bil, J. Effect of Helicobacter Pylori Infection on the Risk of Acute Coronary Syndrome: A Systematic Review and Meta-Analysis. Medecine 2019, 98. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, M.; Zhang, R.; Chen, S.; Xi, Y.; Duan, G. A Meta-Analysis of the Association between Helicobacter Pylori Infection and Risk of Atherosclerotic Cardiovascular Disease. Helicobacter 2020, 25, 12761. [Google Scholar] [CrossRef]
- Gravina, A.G.; Zagari, R.M.; De Musis, C.; Romano, L.; Loguercio, C.; Romano, M. Helicobacter Pylori and Extragastric Diseases: A Review. World J. Gastroenterol. 2018, 24, 3204–3221. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, M.; Miszczyk, E.; Rudnicka, K. Structural Modifications of Helicobacter Pylori Lipopolysaccharide: An Idea for How to Live in Peace. World J. Gastroenterol. 2014, 20, 9882–9897. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A. Predicting the Neuro-Cardio-Haemodynamic Outcomes of Sepsis and Its Pharmacological Interventions: Get to the Future through Numerical Equations. J. Physiol. 2021, 1–3. [Google Scholar] [CrossRef]
- Matsuura, E.; Kobayashi, K.; Matsunami, Y.; Shen, L.; Quan, N.; Makarova, M.; Suchkov, S.V.; Ayada, K.; Oguma, K.; Lopez, L.R. Autoimmunity, Infectious Immunity, and Atherosclerosis. J. Clin. Immunol. 2009, 29, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Farah, R.; Hamza, H.; Khamisy-farah, R. A Link between Platelet to Lymphocyte Ratio and Helicobacter Pylori Infection. J. Clin. Lab. Anal. 2018, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Farah, R.; Khamisy-Farah, R. Association of Neutrophil to Lymphocyte Ratio with Presence and Severity of Gastritis Due to Helicobacter Pylori Infection. J. Clin. Lab. Anal. 2014, 28, 219–223. [Google Scholar] [CrossRef]
- Lee, K.K.; Stelzle, D.; Bing, R.; Anwar, M.; Strachan, F.; Bashir, S.; Newby, D.E.; Shah, J.S.; Chung, M.H.; Bloomfield, G.S.; et al. Global Burden of Atherosclerotic Cardiovascular Disease in People with Hepatitis C Virus Infection: A Systematic Review, Meta-Analysis, and Modelling Study. Lancet Gastroenterol. Hepatol. 2019, 4, 794–804. [Google Scholar] [CrossRef]
- Wen, D.; Du, X.; Dong, J.Z.; Ma, C.S. Hepatitis C Virus Infection and Risk of Coronary Artery Disease: A Meta-Analysis. Eur. J. Intern. Med. 2019, 63, 69–73. [Google Scholar] [CrossRef]
- Babiker, A.; Hassan, M.; Muhammed, S.; Taylor, G.; Poonia, B.; Shah, A.; Bagchi, S. Inflammatory and Cardiovascular Diseases Biomarkers in Chronic Hepatitis C Virus Infection: A Review. Clin. Cardiol. 2020, 43, 222–234. [Google Scholar] [CrossRef]
- Jabeen, S.; Rasheed, A.; Jabeen, N.; Naz, S.A.; Raza, A. Prevalence and Association of HBV and HCV Infection with Cardiovascular Disease Risk Factors in a Peri-Urban Population. J. Pak. Med. Assoc. 2020, 70, 58–63. [Google Scholar] [CrossRef]
- Mehta, D.A.; Cohen, E.; Charafeddine, M.; Cohen, D.E.; Bao, Y.; Sanchez Gonzalez, Y.; Tran, T.T. Effect of Hepatitis C Treatment with Ombitasvir/Paritaprevir/R + Dasabuvir on Renal, Cardiovascular and Metabolic Extrahepatic Manifestations: A Post-Hoc Analysis of Phase 3 Clinical Trials. Infect. Dis. Ther. 2017, 6, 515–529. [Google Scholar] [CrossRef]
- Wernly, B.; Wernly, S.; Niederseer, D.; Datz, C. Hepatitis C Virus (HCV) Infection and Cardiovascular Disease: Hepatologists and Cardiologists Need to Talk! Eur. J. Intern. Med. 2020, 71, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Corrales-Medina, V.F. Association Between Hospitalization for Pneumonia and Subsequent Risk of Cardiovascular Disease. Physiol. Behav. 2014, 63, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Gilley, R.P.; González-Juarbe, N.; Shenoy, A.T.; Reyes, L.F.; Dube, P.H.; Restrepo, M.I.; Orihuela, C.J. Infiltrated Macrophages Die of Pneumolysin-Mediated Necroptosis Following Pneumococcal Myocardial Invasion. Infect. Immun. 2016, 84, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.O.; Millett, E.R.C.; Quint, J.K.; Orihuela, C.J. Cardiotoxicity during Invasive Pneumococcal Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Portier, I.; Campbell, R.A. Role of Platelets in Detection and Regulation of Infection. Arterioscler. Thromb. Vasc. Biol. 2020, 41, 70–78. [Google Scholar] [CrossRef]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The Inflammatory Role of Platelets via Their TLRs and Siglec Receptors. Front. Immunol. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cangemi, R.; Casciaro, M.; Rossi, E.; Calvieri, C.; Bucci, T.; Calabrese, C.M.; Taliani, G.; Falcone, M.; Palange, P.; Bertazzoni, G.; et al. Platelet Activation Is Associated with Myocardial Infarction in Patients with Pneumonia. J. Am. Coll. Cardiol. 2014, 64, 1917–1925. [Google Scholar] [CrossRef]
- Rae, N.; Finch, S.; Chalmers, J.D. Cardiovascular Disease as a Complication of Community-Acquired Pneumonia. Curr. Opin. Pulm. Med. 2016, 22, 212–218. [Google Scholar] [CrossRef]
- Polgreen, L.A.; Riedle, B.N.; Cavanaugh, J.E.; Girotra, S.; London, B.; Schroeder, M.C.; Polgreen, P.M. Estimated Cardiac Risk Associated with Macrolides and Fluoroquinolones Decreases Substantially When Adjusting for Patient Characteristics and Comorbidities. J. Am. Heart Assoc. 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Marra, F.; Zhang, A.; Gillman, E.; Bessai, K.; Parhar, K.; Vadlamudi, N.K. The Protective Effect of Pneumococcal Vaccination on Cardiovascular Disease in Adults: A Systematic Review and Meta-Analysis. Int. J. Infect. Dis. 2020, 99, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus Infection and Relative Risk of Cardiovascular Disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): A Meta-Analysis of Prospective Studies up to 2016. J. Am. Heart Assoc. 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Lv, Y.L.; Han, F.F.; Gong, L.L.; Liu, H.; Ma, J.; Yu, W.Y.; Wan, Z.R.; Jia, Y.J.; Zhang, W.; Shi, M.; et al. Human Cytomegalovirus Infection and Vascular Disease Risk: A Meta-Analysis. Virus Res. 2017, 227, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Freeman, M.L.; Lederman, M.M.; Gianella, S. Partners in Crime: The Role of CMV in Immune Dysregulation and Clinical Outcome During HIV Infection. Curr. HIV/AIDS Rep. 2016, 13, 10–19. [Google Scholar] [CrossRef]
- Nikitskaya, E.; Lebedeva, A.; Ivanova, O.; Maryukhnich, E.; Shpektor, A.; Grivel, J.C.; Margolis, L.; Vasilieva, E. Cytomegalovirus-Productive Infection Is Associated with Acute Coronary Syndrome. J. Am. Heart Assoc. 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.; Sinzger, C. Endothelial Cells in Human Cytomegalovirus Infection: One Host Cell out of Many or a Crucial Target for Virus Spread? Thromb. Haemost. 2009, 102, 1057–1063. [Google Scholar] [CrossRef]
- Weis, M.; Kledal, T.N.; Lin, K.Y.; Panchal, S.N.; Gao, S.Z.; Valantine, H.A.; Mocarski, E.S.; Cooke, J.P. Cytomegalovirus Infection Impairs the Nitric Oxide Synthase Pathway: Role of Asymmetric Dimethylarginine in Transplant Arteriosclerosis. Circulation 2004, 109, 500–505. [Google Scholar] [CrossRef]
- Azevedo, R.B.; Botelho, B.G.; de Hollanda, J.V.G.; Ferreira, L.V.L.; Junqueira de Andrade, L.Z.; Oei, S.S.M.L.; de Souza Mello, T.; Muxfeldt, E.S. Covid-19 and the Cardiovascular System: A Comprehensive Review. J. Hum. Hypertens. 2021, 35, 4–11. [Google Scholar] [CrossRef]
- Szarpak, L.; Filipiak, K.J.; Gasecka, A.; Pruc, M.; Drozd, A.; Jaguszewski, M.J. Correlation between Takotsubo Cardiomyopathy and SARS-CoV-2 Infection. Med. Hypotheses 2021, 146, 110454. [Google Scholar] [CrossRef]
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic Complications in Patients with COVID-19: Pathophysiological Mechanisms, Diagnosis, and Treatment. Cardiovasc. Drugs Ther. 2021, 35, 215–229. [Google Scholar] [CrossRef]
- Zhao, Y.-H.; Zhao, L.; Yang, X.-C.; Wang, P. Cardiovascular Complications of SARS-CoV-2 Infection (COVID-19): A Systematic Review and Meta-Analysis. Rev. Cardiovasc. Med. 2021, 22, 159. [Google Scholar] [CrossRef]
- Gąsecka, A.; Filipiak, K.J.; Jaguszewski, M.J. Impaired Microcirculation Function in Covid-19 and Implications for Potential Therapies. Cardiol. J. 2020, 27, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Szarpak, Ł.; Nowak, B.; Kosior, D.; Zaczynski, A.; Filipiak, K.J.; Jaguszewski, M.J. Cytokines as Predictors of COVID-19 Severity: Evidence from a Meta-Analysis. Polish Arch. Intern. Med. 2021, 131, 98–99. [Google Scholar] [CrossRef]
- Szarpak, L.; Zaczynski, A.; Kosior, D.; Bialka, S.; Ladny, J.R.; Gilis-Malinowska, N.; Smereka, J.; Kanczuga-Koda, L.; Gasecka, A.; Filipiak, K.J.; et al. Evidence of Diagnostic Value of Ferritin in Patients with COVID-19. Cardiol. J. 2020, 27, 886–887. [Google Scholar] [CrossRef]
- Hertanto, D.M.; Sutanto, H.; Wungu, C.D.K. Immunomodulation as a Potent COVID-19 Pharmacotherapy: Past, Present and Future. Preprints 2021. [Google Scholar] [CrossRef]
- Evans, P.C.; Ed Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial Dysfunction in COVID-19: A Position Paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Ruetzler, K.; Szarpak, L.; Ladny, J.R.; Gasecka, A.; Malinowska, N.G.; Pruc, M.; Smereka, J.; Nowak, B.; Filipiak, K.J.; Jaguszewski, M.J. D-Dimer Levels Predict COVID-19 Severity and Mortality. Kardiol. Pol. 2021, 79, 217–219. [Google Scholar] [CrossRef]
- Long, B.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Cardiovascular complications in COVID-19. Am. J. Emerg. Med. 2020, 38, 1504–1507. [Google Scholar] [CrossRef]
- Gasecka, A.; Pruc, M.; Kukula, K.; Gilis-Malinowska, N.; Filipiak, K.J.; Jaguszewski, M.J.; Szarpak, L. Post-Covid-19 Heart Syndrome. Cardiol. J. 2021, 28, 353–354. [Google Scholar] [CrossRef]
- Freiberg, M.S.; Chang, C.H.; Kuller, L.H.; Goetz, M.B.; Leaf, D.; Oursler, K.A.; Mcginnis, K.; Crothers, K.; Sico, J.; Crane, H.; et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern. Med. 2013, 173, 614–622. [Google Scholar] [CrossRef] [PubMed]
- So-Armah, K.; Benjamin, L.A.; Bloomfield, G.S.; Feinstein, M.J.; Hsue, P.; Njuguna, B.; Freiberg, M.S. HIV and Cardiovascular Disease. Lancet HIV 2020, 7, E279–E293. [Google Scholar] [CrossRef]
- Visser, M.R.; Vercellotti, G.M. Herpes Simplex Virus and Atherosclerosis. Eur. Heart J. 1993, 14 (SuppL K), 39–42. [Google Scholar]
- Vilkuna-Rautiainen, T.; Pussinen, P.J.; Roivainen, M.; Petäys, T.; Jousilahti, P.; Hovi, T.; Vartiainen, E.; Asikainen, S. Serum Antibody Response to Periodontal Pathogens and Herpes Simplex Virus in Relation to Classic Risk Factors of Cardiovascular Disease. Int. J. Epidemiol. 2006, 35, 1486–1494. [Google Scholar] [CrossRef]
- Mendy, A.; Vieira, E.R.; Gasana, J. Seropositivity to Herpes Simplex Virus Type 2, but Not Type 1 Is Associated with Premature Cardiovascular Diseases: A Population-Based Cross-Sectional Study. Atherosclerosis 2013, 231, 18–21. [Google Scholar] [CrossRef]
- Rosenfeld, M.E.; Campbell, L.A. Pathogens and Atherosclerosis: Update on the Potential Contribution of Multiple Infectious Organisms to the Pathogenesis of Atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef]
- Pothineni, N.V.K.; Subramany, S.; Kuriakose, K.; Shirazi, L.F.; Romeo, F.; Shah, P.K.; Mehta, J.L. Infections, Atherosclerosis, and Coronary Heart Disease. Eur. Heart J. 2017, 38, 3195–3201. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Fonseca, F.A.; Genest, J.; Gotto, A.M.; Kastelein, J.J.; Koenig, W.; Libby, P.; Lorenzatti, A.J.; MacFadyen, J.G.; et al. Reduction in C-Reactive Protein and LDL Cholesterol and Cardiovascular Event Rates after Initiation of Rosuvastatin: A Prospective Study of the JUPITER Trial. Lancet 2009, 373, 1175–1182. [Google Scholar] [CrossRef]
- Thomas, M.R.; Outteridge, S.N.; Ajjan, R.A.; Phoenix, F.; Sangha, G.K.; Faulkner, R.E.; Ecob, R.; Judge, H.M.; Khan, H.; West, L.E.; et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and Its Prothrombotic Effects in an Experimental Human Model. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2562–2570. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.; Solomon, S.; Pieper, K.; Reed, S.; Rouleau, J.; Velazquez, E.; White, H.; Howlett, J.; Swedberg, K.; Maggioni, A.; et al. The Effect of Valsartan, Captopril, or Both on Atherosclerotic Events after Acute Myocardial Infarction: An Analysis of the Valsartan in Acute Myocardial Infarction Trial (VALIANT). J. Am. Coll. Cardiol. 2006, 47, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Greenberg, J.D.; Kremer, J.M.; Curtis, J.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Farkouh, M.E.; Nasir, A.; Setoguchi, S.; Solomon, D.H. Tumour Necrosis Factor Antagonist Use and Associated Risk Reduction of Cardiovascular Events among Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 2011, 70, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; Macfadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Heart, N.; Rosenberg, Y.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Welsh, P.; Grassia, G.; Botha, S.; Sattar, N.; Maffia, P. Targeting Inflammation to Reduce Cardiovascular Disease Risk: A Realistic Clinical Prospect? Br. J. Pharmacol. 2017, 174, 3898–3913. [Google Scholar] [CrossRef]
- Pradhan, A.D.; Aday, A.W.; Rose, L.M.; Ridker, P.M. Residual Inflammatory Risk on Treatment with PCSK9 Inhibition and Statin Therapy. Circulation 2018, 138, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Tokgözoğlu, L.; Corsini, A.; Sirtori, C.R. PCSK9 Inhibition and Inflammation: A Narrative Review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef]
- Pertzov, B.; Eliakim-Raz, N.; Atamna, H.; Trestioreanu, A.Z.; Yahav, D.; Leibovici, L. Hydroxymethylglutaryl-CoA Reductase Inhibitors (Statins) for the Treatment of Sepsis in Adults—A Systematic Review and Meta-Analysis. Clin. Microbiol. Infect. 2019, 25, 280–289. [Google Scholar] [CrossRef]
- Lowenstern, A.; Storey, R.F.; Neely, M.; Sun, J.L.; Angiolillo, D.J.; Cannon, C.P.; Himmelmann, A.; Huber, K.; James, S.K.; Katus, H.A.; et al. Platelet-Related Biomarkers and Their Response to Inhibition with Aspirin and P2y12-Receptor Antagonists in Patients with Acute Coronary Syndrome. J. Thromb. Thrombolysis 2017, 44, 145–153. [Google Scholar] [CrossRef]
- Husted, S.; Storey, R.F.; Harrington, R.A.; Emanuelsson, H.; Cannon, C.P. Changes in Inflammatory Biomarkers in Patients Treated with Ticagrelor or Clopidogrel. Clin. Cardiol. 2010, 33, 206–212. [Google Scholar] [CrossRef]
- Kiers, D.; van der Heijden, W.A.; van Ede, L.; Gerretsen, J.; de Mast, Q.; van der Ven, A.J.; El Messaoudi, S.; Rongen, G.A.; Gomes, M.; Kox, M.; et al. A Randomized Trial on the Effect of Anti-Platelet Therapy on the Systemic Inflammatory Response in Human Endotoxemia. Thrombo. Haemost. 2017, 117, 1798–1807. [Google Scholar] [CrossRef]
- Gasecka, A.; Nieuwland, R.; Budnik, M.; Dignat-George, F.; Eyileten, C.; Harrison, P.; Lacroix, R.; Leroyer, A.; Opolski, G.; Pluta, K.; et al. Ticagrelor Attenuates the Increase of Extracellular Vesicle Concentrations in Plasma after Acute Myocardial Infarction Compared to Clopidogrel. J. Thromb. Haemost. 2020, 18, 609–623. [Google Scholar] [CrossRef]
- Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tikellis, C.; Thomas, M.C.; Golledge, J. Angiotensin Converting Enzyme 2 and Atherosclerosis. Atherosclerosis 2013, 226, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Ranjbar, R.; Shafiee, M.; Hesari, A.R.; Ferns, G.A.; Ghasemi, F.; Avan, A. The Potential Therapeutic Use of Renin–Angiotensin System Inhibitors in the Treatment of Inflammatory Diseases. J. Cell. Physiol. 2019, 234, 2277–2295. [Google Scholar] [CrossRef]
- Mitrovic, V.; Klein, H.H.; Krekel, N.; Kreuzer, J.; Fichtlscherer, S.; Schirmer, A.; Paar, W.D.; Hamm, C.W. Influence of the Angiotensin Converting Enzyme Inhibitor Ramipril on High-Sensitivity C-Reactive Protein (Hs-CRP) in Patients with Documented Atherosclerosis. Z. Kardiol. 2005, 94, 336–342. [Google Scholar] [CrossRef]
- Ceconi, C.; Fox, K.M.; Remme, W.J.; Simoons, M.L.; Deckers, J.W.; Bertrand, M.; Parrinello, G.; Kluft, C.; Blann, A.; Cokkinos, D.; et al. ACE Inhibition with Perindopril and Biomarkers of Atherosclerosis and Thrombosis: Results from the PERTINENT Study. Atherosclerosis 2009, 204, 273–275. [Google Scholar] [CrossRef]
- Han, S.H.; Chung, W.J.; Kang, W.C.; Lee, K.; Park, Y.M.; Shin, M.S.; Ahn, T.H.; Choi, I.S.; Shin, E.K. Rosuvastatin Combined with Ramipril Significantly Reduced Atheroma Volume by Anti-Inflammatory Mechanism: Comparative Analysis with Rosuvastatin Alone by Intravascular Ultrasound. Int. J. Cardiol. 2012, 158, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Martínez, G.J.; Robertson, S.; Barraclough, J.; Xia, Q.; Mallat, Z.; Bursill, C.; Celermajer, D.S.; Patel, S. Colchicine Acutely Suppresses Local Cardiac Production of Inflammatory Cytokines in Patients with an Acute Coronary Syndrome. J. Am. Heart Assoc. 2015, 4, e002128. [Google Scholar] [CrossRef]
- Kajikawa, M.; Higashi, Y.; Tomiyama, H.; Maruhashi, T.; Kurisu, S.; Kihara, Y.; Mutoh, A.; Ueda, S. ichiro. Effect of Short-Term Colchicine Treatment on Endothelial Function in Patients with Coronary Artery Disease. Int. J. Cardiol. 2019, 281, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Kurup, R.; Barraclough, J.; Henriquez, R.; Cartland, S.; Arnott, C.; Misra, A.; Martínez, G.; Kavurma, M.; Patel, S. Colchicine as a Novel Therapy for Suppressing Chemokine Production in Patients with an Acute Coronary Syndrome: A Pilot Study. Clin. Ther. 2019, 41, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, A.T.L.; Nidorf, S.M.; Mosterd, A.; Cornel, J.H. Colchicine in Stable Coronary Artery Disease. Clin. Ther. 2019, 41, 30–40. [Google Scholar] [CrossRef] [PubMed]
- McKnight, A.H.; Katzenberger, D.R.; Britnell, S.R. Colchicine in Acute Coronary Syndrome: A Systematic Review. Ann. Pharmacother. 2021, 55, 187–197. [Google Scholar] [CrossRef]
- Harrington, R.A. Targeting Inflammation in Coronary Artery Disease. N. Engl. J. Med. 2017, 377, 1197–1198. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Authors | Therapy | Mechanism of Action | Information about Study | Outcomes | Effect |
|---|---|---|---|---|---|
| Ridker et al., 2009 [111] | Rosuvastatin vs. placebo | HMG-CoA inhibitor, pleiotropic effects | A randomized, double-blind, placebo-controlled trial including 15,548 initially healthy men and women | Cardiovascular death, non-fatal stroke, non-fatal AMI, hospitalization due to unstable angina, revascularization | ↓ risk of adverse outcomes (HR = 0.35; 95% CI: 0.23–0.54; p < 0.0001) |
| Thomas et al., 2015 [112] | Ticagrelor vs. clopidogrel vs. placebo | Inhibition of P2Y12 receptor | Randomized injection of E. coli endotoxins to 30 healthy volunteers (10-ticagrelor, 10-clopidogrel, 10-placeboes) | Concentrations of inflammatory biomarkers | Ticagrelor and clopidogrel: ↓ IL6, TNF-α, CCL2 Only ticagrelor: ↓ G-CSF, IL-8; ↑ IL-10; ↔ hsCRP |
| McMurray et al., 2006 [113] | Valsartan vs. captopril | ARB or ACE inhibition | Randomized 14,703 high-risk patients with acute MI to receive captopril or valsartan or the combination of the two | All-cause mortality, cardiovascular mortality, non-fatal cardiovascular events | ↓ risk of adverse outcomes; similar effect of ARBs and ACE-I (HR = 0.97; 95% CI:0.91–1.03; p = 0.286) |
| Tardif et al., 2020 [114] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, double-blind, placebo-controlled trial including 4745 patients with recent AMI (~2 weeks before) | Cardiovascular death, resuscitated cardiac arrest, AMI, stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.77; 95% CI: 0.61–0.96; p = 0.02) |
| Nidorf et al., 2019 [115] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A randomized, placebo-controlled, double-blind trial including 5522 patients with chronic coronary syndrome | Cardiovascular death, MI, ischemic stroke, coronary revascularization | ↓ risk of adverse outcomes (HR = 0.69; 95% CI: 0.57–0.83; p < 0.001) |
| Nidorf et al., 2013 [116] | Colchicine 0.5 mg daily vs. placebo | NLRP3 inflammasome inhibitor | A prospective, randomized, observer-blinded, placebo-controlled clinical trial including 532 patients with stable coronary disease | Acute coronary syndrome, out-of-hospital cardiac arrest, ischemic stroke | ↓ risk of adverse outcomes (HR = 0.33; 95% CI: 0.18–0.59; p < 0.001) |
| Ridker et al., 2017 [117] | Canakinumab 150 mg every 3 months vs. placebo | Monoclonal anti-IL-1β antibody | A randomized, double-blind, placebo-controlled trial including 10,061 patients with previous AMI and hsCRP ≥ 2 mg/L | Non-fatal myocardial infarction, nonfatal stroke, cardiovascular death | ↓ risk of adverse outcomes HR = 0.85 (95% CI: 0.74–0.98; p = 0.021) |
| Greenberg et al., 2010 [118] | TNF-α antagonists vs. DMARDs | TNF-α inhibition | A longitudinal cohort study of 10,156 rheumatoid arthritis patients enrolled in the US-based CORRONA database | Non-fatal MI, transient ischemic attack, stroke, cardiovascular death | ↓ risk of adverse outcomes by TNF-α (HR = 0.39; 95% CI 0.19–0.82) |
| Ridker et al., 2019 [119] | Methotrexate 15–20 mg/week vs. placebo | Antimetabolite, immune-system suppressant | A randomized, double-blind, placebo-controlled trial including 4786 patients with previous MI or multivessel coronary disease, additionally with type 2 diabetes or metabolic syndrome | Nonfatal MI, nonfatal stroke, cardiovascular death, unstable angina | ↔ adverse outcomes (HR = 0.96; 95% CI: 0.79–1.16; p = 0.67) ↔ hsCRP, IL-1β, IL-6 ↑ ALT, AST |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szwed, P.; Gąsecka, A.; Zawadka, M.; Eyileten, C.; Postuła, M.; Mazurek, T.; Szarpak, Ł.; Filipiak, K.J. Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications. J. Clin. Med. 2021, 10, 2539. https://doi.org/10.3390/jcm10122539
Szwed P, Gąsecka A, Zawadka M, Eyileten C, Postuła M, Mazurek T, Szarpak Ł, Filipiak KJ. Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications. Journal of Clinical Medicine. 2021; 10(12):2539. https://doi.org/10.3390/jcm10122539
Chicago/Turabian StyleSzwed, Piotr, Aleksandra Gąsecka, Mateusz Zawadka, Ceren Eyileten, Marek Postuła, Tomasz Mazurek, Łukasz Szarpak, and Krzysztof J. Filipiak. 2021. "Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications" Journal of Clinical Medicine 10, no. 12: 2539. https://doi.org/10.3390/jcm10122539
APA StyleSzwed, P., Gąsecka, A., Zawadka, M., Eyileten, C., Postuła, M., Mazurek, T., Szarpak, Ł., & Filipiak, K. J. (2021). Infections as Novel Risk Factors of Atherosclerotic Cardiovascular Diseases: Pathophysiological Links and Therapeutic Implications. Journal of Clinical Medicine, 10(12), 2539. https://doi.org/10.3390/jcm10122539