
Anti-Inflammatory Drugs in Patients with Ischemic Heart Disease

 ,
,  , and
, and 
Abstract
:1. Introduction
1.1. Atheroma Plaque Formation and Its Inflammatory Components
1.2. Relationship between Pro-Inflammatory States and Acute Coronary Events
1.3. Inflammatory Biomarkers and Atherosclerosis
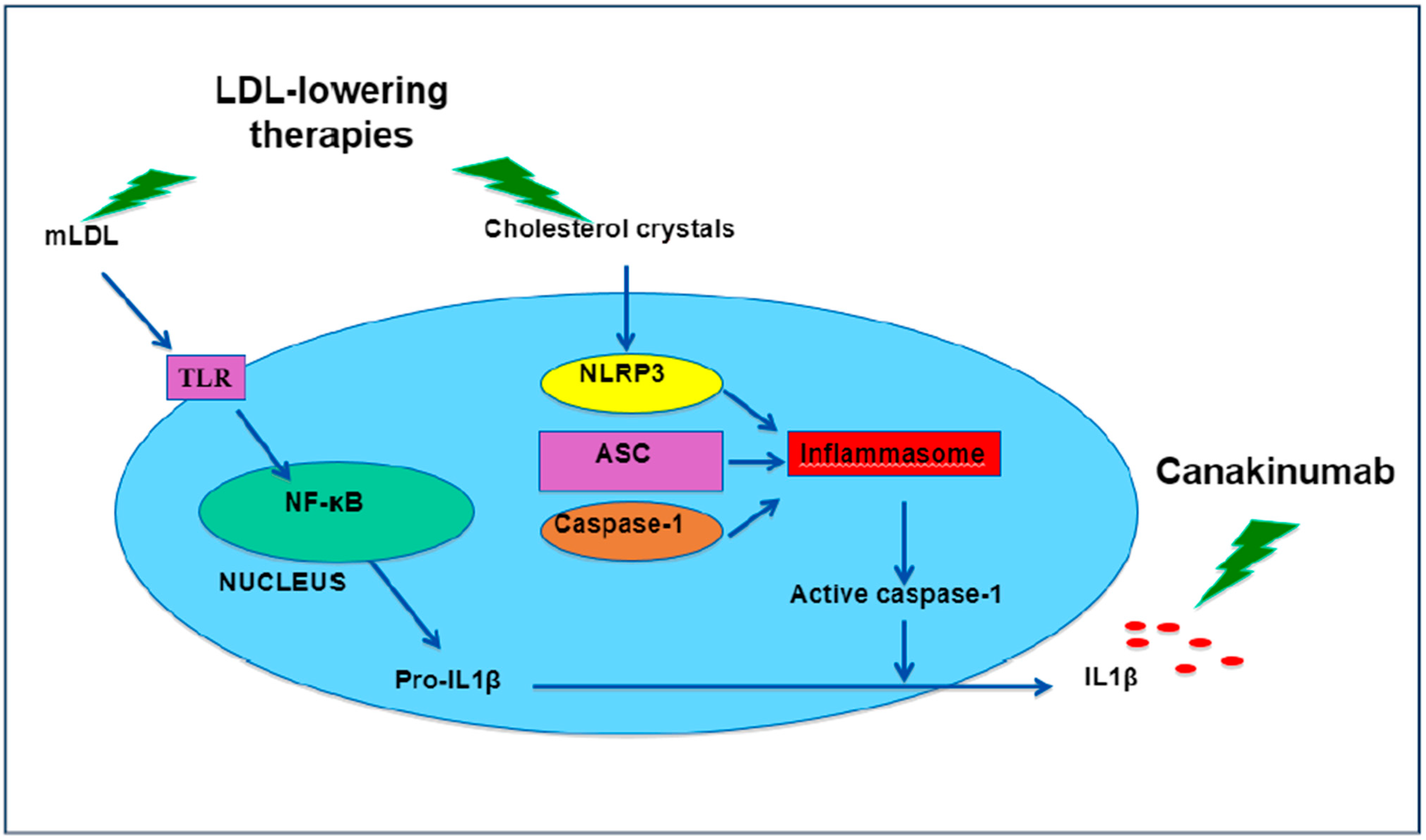
2. Drugs Used in Therapy for Atherosclerotic Disease Reduce Inflammation
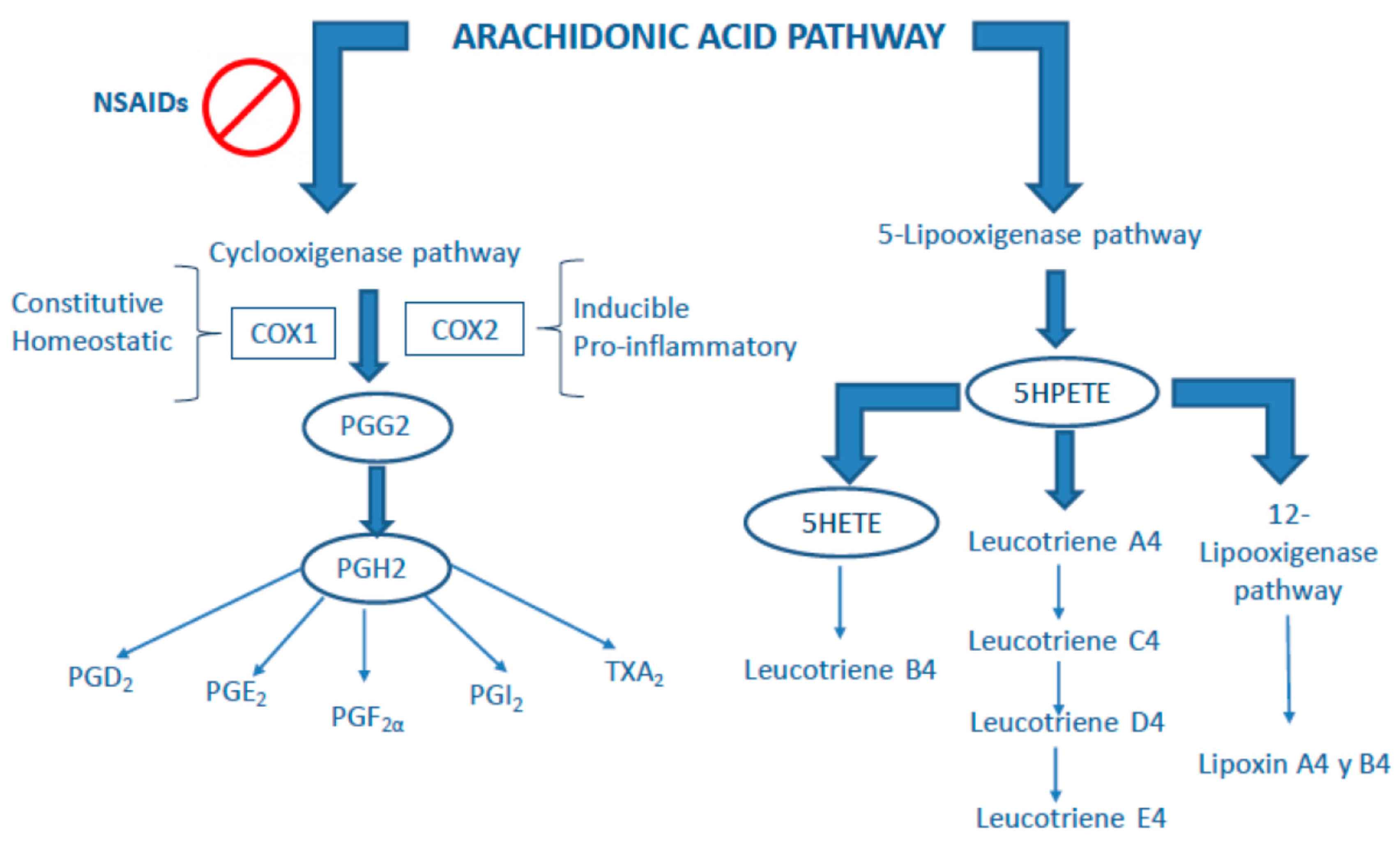
3. Classical Anti-Inflammatory Drugs
4. Non-Classical Anti-Inflammatory Drugs
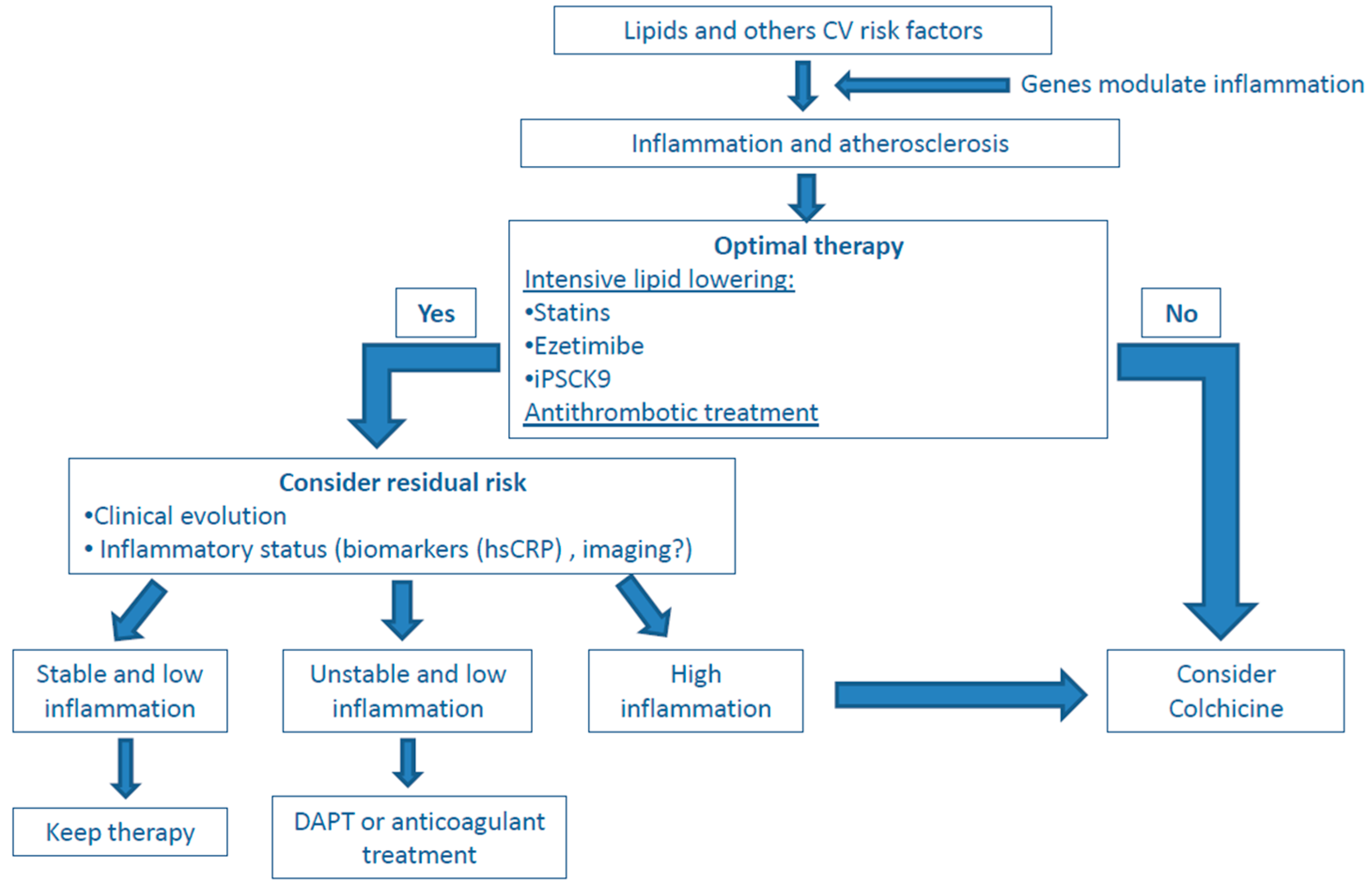
5. Present and Future Role of Anti-Inflammatory Drugs in Atherosclerosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Henry, P.D.; Chen, C.H. Inflammatory mechanisms of atheroma formation. Influence of fluid mechanics and lipid-derived inflammatory mediators. Am. J. Hypertens. 1993, 6, 328S–334S. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.B.; Nordestgaard, B.G.; Stender, S.; Kjeldsen, K. Aortic permeability to LDL as a predictor of aortic cholesterol accumulation in cholesterol-fed rabbits. Arterioscler. Thromb. 1992, 12, 1402–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; La Fata, V.; Plutzky, J.; Liao, J.K. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998, 97, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [Green Version]
- Camejo, G.; Hurt-Camejo, E.; Wiklund, O.; Bondjers, G. Association of apo B lipoproteins with arterial proteoglycans: Pathological significance and molecular basis. Atherosclerosis 1998, 139, 205–222. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Robbesyn, F.; Garcia, V.; Auge, N.; Vieira, O.; Frisach, M.-F.; Salvayre, R.; Negre-Salvayre, A. HDL counterbalance the proinflammatory effect of oxidized LDL by inhibiting intracellular reactive oxygen species rise, proteasome activation, and subsequent NF-kappaB activation in smooth muscle cells. FASEB J. 2003, 17, 743–745. [Google Scholar] [CrossRef] [Green Version]
- Nelken, N.A.; Coughlin, S.R.; Gordon, D.; Wilcox, J.N. Monocyte chemoattractant protein-1 in human atheromatous plaques. J. Clin. Investig. 1991, 88, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Egashira, K.; Ni, W.; Kitamoto, S.; Usui, M.; Otani, K.; Ishibashi, M.; Hiasa, K.; Nishida, K.; Takeshita, A. Anti-monocyte chemoattractant protein-1 gene therapy limits progression and destabilization of established atherosclerosis in apolipoprotein E-knockout mice. Circulation 2002, 106, 2700–2706. [Google Scholar] [CrossRef]
- Cyrus, T.; Witztum, J.L.; Rader, D.J.; Tangirala, R.; Fazio, S.; Linton, M.F.; Funk, C.D. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Investig. 1999, 103, 1597–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyy, Y.J.; Hsieh, H.J.; Usami, S.; Chien, S. Fluid shear stress induces a biphasic response of human monocyte chemotactic protein 1 gene expression in vascular endothelium. Proc. Natl. Acad. Sci. USA 1994, 91, 4678–4682. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M.; Neumann, F.J.; Dickfeld, T.; Koch, W.; Laugwitz, K.L.; Adelsberger, H.; Langenbrink, K.; Page, S.; Neumeier, D.; Schömig, A.; et al. Activated platelets induce monocyte chemotactic protein-1 secretion and surface expression of intercellular adhesion molecule-1 on endothelial cells. Circulation 1998, 98, 1164–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, S.M.; deBlois, D.; O’Brien, E.R. The intima. Soil for atherosclerosis and restenosis. Circ. Res. 1995, 77, 445–465. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef]
- Tuñón, J.; Ruiz-Ortega, M.; Egido, J. Regulation of matrix proteins and impact on vascular structure. Curr. Hypertens. Rep. 2000, 2, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Oemar, B.S.; Werner, A.; Garnier, J.M.; Do, D.D.; Godoy, N.; Nauck, M.; März, W.; Rupp, J.; Pech, M.; Lüscher, T.F. Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation 1997, 95, 831–839. [Google Scholar] [CrossRef]
- O’Brien, E.R.; Garvin, M.R.; Dev, R.; Stewart, D.K.; Hinohara, T.; Simpson, J.B.; Schwartz, S.M. Angiogenesis in human coronary atherosclerotic plaques. Am. J. Pathol. 1994, 145, 883–894. [Google Scholar]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 2003, 349, 2316–2325. [Google Scholar] [CrossRef]
- Rekhter, M.D.; Zhang, K.; Narayanan, A.S.; Phan, S.; Schork, M.A.; Gordon, D. Type I collagen gene expression in human atherosclerosis. Localization to specific plaque regions. Am. J. Pathol. 1993, 143, 1634–1648. [Google Scholar] [PubMed]
- Cheng, G.C.; Loree, H.M.; Kamm, R.D.; Fishbein, M.C.; Lee, R.T. Distribution of circumferential stress in ruptured and stable atherosclerotic lesions. A structural analysis with histopathological correlation. Circulation 1993, 87, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Martín-Ventura, J.L.; Blanco-Colio, L.M.; Muñoz-García, B.; Gómez-Hernández, A.; Arribas, A.; Ortega, L.; Tuñón, J.; Egido, J. NF-kappaB activation and Fas ligand overexpression in blood and plaques of patients with carotid atherosclerosis: Potential implication in plaque instability. Stroke 2004, 35, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.H.; Best, P.J.M.; Edwards, W.D.; Holmes, D.R.; Carlson, P.J.; Celermajer, D.S.; Lerman, A. Nuclear factor-kappaB immunoreactivity is present in human coronary plaque and enhanced in patients with unstable angina pectoris. Atherosclerosis 2002, 160, 147–153. [Google Scholar] [CrossRef]
- van der Wal, A.C.; Becker, A.E.; van der Loos, C.M.; Das, P.K. Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation 1994, 89, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Moreno, P.R.; Falk, E.; Palacios, I.F.; Newell, J.B.; Fuster, V.; Fallon, J.T. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation 1994, 90, 775–778. [Google Scholar] [CrossRef] [Green Version]
- Falk, E.; Shah, P.K.; Fuster, V. Coronary plaque disruption. Circulation 1995, 92, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Roitman-Johnson, B.; Stampfer, M.J.; Allen, J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet 1998, 351, 88–92. [Google Scholar] [CrossRef]
- Kai, H.; Ikeda, H.; Yasukawa, H.; Kai, M.; Seki, Y.; Kuwahara, F.; Ueno, T.; Sugi, K.; Imaizumi, T. Peripheral blood levels of matrix metalloproteases-2 and -9 are elevated in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 1998, 32, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Georgakis, M.K.; van der Laan, S.W.; Asare, Y.; Mekke, J.M.; Haitjema, S.; Schoneveld, A.H.; de Jager, S.C.A.; Nurmohamed, N.S.; Kroon, J.; Stroes, E.S.G.; et al. Monocyte-Chemoattractant Protein-1 Levels in Human Atherosclerotic Lesions Associate With Plaque Vulnerability. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- de Lemos, J.A.; Morrow, D.A.; Sabatine, M.S.; Murphy, S.A.; Gibson, C.M.; Antman, E.M.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation 2003, 107, 690–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuñón, J.; Blanco-Colio, L.; Cristóbal, C.; Tarín, N.; Higueras, J.; Huelmos, A.; Alonso, J.; Egido, J.; Asensio, D.; Lorenzo, Ó.; et al. Usefulness of a combination of monocyte chemoattractant protein-1, galectin-3, and N-terminal probrain natriuretic peptide to predict cardiovascular events in patients with coronary artery disease. Am. J. Cardiol. 2014, 113, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Colio, L.M.; Méndez-Barbero, N.; Pello Lázaro, A.M.; Aceña, Á.; Tarín, N.; Cristóbal, C.; Martínez-Milla, J.; González-Lorenzo, Ó.; Martín-Ventura, J.L.; Huelmos, A.; et al. MCP-1 Predicts Recurrent Cardiovascular Events in Patientswith Persistent Inflammation. J. Clin. Med. 2021, 10, 1137. [Google Scholar] [CrossRef]
- André, P.; Prasad, K.S.S.; Denis, C.V.; He, M.; Papalia, J.M.; Hynes, R.O.; Phillips, D.R.; Wagner, D.D. CD40L stabilizes arterial thrombi by a beta3 integrin--dependent mechanism. Nat. Med. 2002, 8, 247–252. [Google Scholar] [CrossRef]
- Solano-López, J.; Zamorano, J.L.; Pardo Sanz, A.; Amat-Santos, I.; Sarnago, F.; Gutiérrez Ibañes, E.; Sanchis, J.; Rey Blas, J.R.; Gómez-Hospital, J.A.; Santos Martínez, S.; et al. Risk factors for in-hospital mortality in patients with acute myocardial infarction during the COVID-19 outbreak. Rev. Esp. Cardiol. 2020, 73, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Choudry, F.A.; Hamshere, S.M.; Rathod, K.S.; Akhtar, M.M.; Archbold, R.A.; Guttmann, O.P.; Woldman, S.; Jain, A.K.; Knight, C.J.; Baumbach, A.; et al. High Thrombus Burden in Patients With COVID-19 Presenting With ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 76, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cushman, M.; Stampfer, M.J.; Tracy, R.P.; Hennekens, C.H. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N. Engl. J. Med. 1997, 336, 973–979. [Google Scholar] [CrossRef]
- Emberson, J.; Bennett, D.; Link, E.; Parish, S.; Danesh, J.; Armitage, J.; Collins, R. C-reactive protein concentration and the vascular benefits of statin therapy: An analysis of 20,536 patients in the Heart Protection Study. Lancet 2011, 377, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Piepoli, M.F.; Hoes, A.W.; Agewall, S.; Albus, C.; Brotons, C.; Catapano, A.L.; Cooney, M.-T.; Corrà, U.; Cosyns, B.; Deaton, C.; et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representati. Eur. Heart J. 2016, 37, 2315–2381. [Google Scholar] [CrossRef]
- Goff, D.C.J.; Lloyd-Jones, D.M.; Bennett, G.; Coady, S.; D’Agostino, R.B.; Gibbons, R.; Greenland, P.; Lackland, D.T.; Levy, D.; O’Donnell, C.J.; et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S49–S73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herder, C.; Baumert, J.; Thorand, B.; Martin, S.; Löwel, H.; Kolb, H.; Koenig, W. Chemokines and incident coronary heart disease: Results from the MONICA/KORA Augsburg case-cohort study, 1984–2002. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2147–2152. [Google Scholar] [CrossRef] [Green Version]
- Georgakis, M.K.; de Lemos, J.A.; Ayers, C.; Wang, B.; Björkbacka, H.; Pana, T.A.; Thorand, B.; Sun, C.; Fani, L.; Malik, R.; et al. Association of Circulating Monocyte Chemoattractant Protein-1 Levels With Cardiovascular Mortality: A Meta-analysis of Population-Based Studies. JAMA Cardiol. 2021, 6, 587–592. [Google Scholar] [CrossRef]
- Younus, A.; Humayun, C.; Ahmad, R.; Ogunmoroti, O.; Kandimalla, Y.; Aziz, M.; Malik, R.; Saand, A.R.; Valdes, C.; Badlani, R.; et al. Lipoprotein-associated phospholipase A2 and its relationship with markers of subclinical cardiovascular disease: A systematic review. J. Clin. Lipidol. 2017, 11, 328–337. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-Associated Phospholipase A2 Protein Expression in the Natural Progression of Human Coronary Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weintraub, H.S. Identifying the Vulnerable Patient with Rupture-Prone Plaque. Am. J. Cardiol. 2008, 101, S3–S10. [Google Scholar] [CrossRef]
- Packard, C.J.; O’Reilly, D.S.J.; Caslake, M.J.; McMahon, A.D.; Ford, I.; Cooney, J.; Macphee, C.H.; Suckling, K.E.; Krishna, M.; Wilkinson, F.E.; et al. Lipoprotein-Associated Phospholipase A2 as an Independent Predictor of Coronary Heart Disease. N. Engl. J. Med. 2000, 343, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Lp-PLA2 Studies Collaboration. Lipoprotein-associated phospholipase A2 and risk of coronary disease, stroke, and mortality: Collaborative analysis of 32 prospective studies. Lancet 2010, 375, 1536–1544. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.J.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, T.; Sugiyama, S.; Koga, H.; Sugamura, K.; Ohba, K.; Matsuzawa, Y.; Sumida, H.; Matsui, K.; Jinnouchi, H.; Ogawa, H. Significance of a multiple biomarkers strategy including endothelial dysfunction to improve risk stratification for cardiovascular events in patients at high risk for coronary heart disease. J. Am. Coll. Cardiol. 2009, 54, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, H.C.; Divya, A.; Prasad, J.; Mittal, A. Plasma circulatory markers in male and female patients with coronary artery disease. Heart Lung 2010, 39, 296–303. [Google Scholar] [CrossRef]
- Leite, A.R.; Borges-Canha, M.; Cardoso, R.; Neves, J.S.; Castro-Ferreira, R.; Leite-Moreira, A. Novel Biomarkers for Evaluation of Endothelial Dysfunction. Angiology 2020, 71, 397–410. [Google Scholar] [CrossRef]
- Blanco-Colio, L.M. TWEAK/Fn14 Axis: A Promising Target for the Treatment of Cardiovascular Diseases. Front. Immunol. 2014, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Boekholdt, S.M.; Peters, R.J.G.; Hack, C.E.; Day, N.E.; Luben, R.; Bingham, S.A.; Wareham, N.J.; Reitsma, P.H.; Khaw, K.-T. IL-8 plasma concentrations and the risk of future coronary artery disease in apparently healthy men and women: The EPIC-Norfolk prospective population study. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1503–1508. [Google Scholar] [CrossRef] [Green Version]
- Jefferis, B.J.M.H.; Papacosta, O.; Owen, C.G.; Wannamethee, S.G.; Humphries, S.E.; Woodward, M.; Lennon, L.T.; Thomson, A.; Welsh, P.; Rumley, A.; et al. Interleukin 18 and coronary heart disease: Prospective study and systematic review. Atherosclerosis 2011, 217, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Tang, Q.; Zhu, X.; Yang, X. IL-37 increased in patients with acute coronary syndrome and associated with a worse clinical outcome after ST-segment elevation acute myocardial infarction. Clin. Chim. Acta. 2017, 468, 140–144. [Google Scholar] [CrossRef]
- Held, C.; White, H.D.; Stewart, R.A.H.; Budaj, A.; Cannon, C.P.; Hochman, J.S.; Koenig, W.; Siegbahn, A.; Steg, P.G.; Soffer, J.; et al. Inflammatory Biomarkers Interleukin-6 and C-Reactive Protein and Outcomes in Stable Coronary Heart Disease: Experiences From the STABILITY (Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) Trial. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Erl, W.; Pietsch, A.; Weber, P.C. Aspirin inhibits nuclear factor-kappa B mobilization and monocyte adhesion in stimulated human endothelial cells. Circulation 1995, 91, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation. Eur. Heart J. 2020. [Google Scholar] [CrossRef]
- Hernández-Presa, M.; Bustos, C.; Ortego, M.; Tuñon, J.; Renedo, G.; Ruiz-Ortega, M.; Egido, J. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-kappa B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997, 95, 1532–1541. [Google Scholar] [CrossRef]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of angiotensin II and interleukin 6 in human coronary atherosclerotic plaques: Potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef] [Green Version]
- Tuñón, J.; Badimón, L.; Bochaton-Piallat, M.-L.; Cariou, B.; Daemen, M.J.; Egido, J.; Evans, P.C.; Hoefer, I.E.; Ketelhuth, D.F.J.; Lutgens, E.; et al. Identifying the anti-inflammatory response to lipid lowering therapy: A position paper from the working group on atherosclerosis and vascular biology of the European Society of Cardiology. Cardiovasc. Res. 2019, 115, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Scandinavian Simvastatin Survival Study Group. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: The Scandinavian Simvastatin Survival Study (4S). Lancet 1994, 344, 1383–1389. [Google Scholar] [CrossRef]
- Shepherd, J.; Cobbe, S.M.; Ford, I.; Isles, C.G.; Lorimer, A.R.; MacFarlane, P.W.; McKillop, J.H.; Packard, C.J.; West of Scotland Coronary Prevention Study Group. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. 1995. Atheroscler. Suppl. 2004, 5, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bustos, C.; Hernández-Presa, M.A.; Ortego, M.; Tuñón, J.; Ortega, L.; Pérez, F.; Díaz, C.; Hernández, G.; Egido, J. HMG-CoA reductase inhibition by atorvastatin reduces neointimal inflammation in a rabbit model of atherosclerosis. J. Am. Coll. Cardiol. 1998, 32, 2057–2064. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Hernández, A.; Sánchez-Galán, E.; Martín-Ventura, J.L.; Vidal, C.; Blanco-Colio, L.M.; Ortego, M.; Vega, M.; Serrano, J.; Ortega, L.; Hernández, G.; et al. Atorvastatin reduces the expression of prostaglandin E2 receptors in human carotid atherosclerotic plaques and monocytic cells: Potential implications for plaque stabilization. J. Cardiovasc. Pharmacol. 2006, 47, 60–69. [Google Scholar] [CrossRef]
- Azar, R.R.; Rinfret, S.; Théroux, P.; Stone, P.H.; Dakshinamurthy, R.; Feng, Y.J.; Wu, A.H.; Rangé, G.; Waters, D.D. A randomized placebo-controlled trial to assess the efficacy of antiinflammatory therapy with methylprednisolone in unstable angina (MUNA trial). Eur. Heart J. 2000, 21, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.; Henry, D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: Systematic review of population-based controlled observational studies. PLoS Med. 2011, 8, e1001098. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Yeomans, N.D.; Solomon, D.H.; Lüscher, T.F.; Libby, P.; Husni, M.E.; Graham, D.Y.; Borer, J.S.; Wisniewski, L.M.; Wolski, K.E.; et al. Cardiovascular Safety of Celecoxib, Naproxen, or Ibuprofen for Arthritis. N. Engl. J. Med. 2016, 375, 2519–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, P.L.; Brown, E.J.; Gunther, S.; Alexander, R.W.; Barry, W.H.; Mudge, G.H.; Grossman, W. Coronary vasoconstrictor effect of indomethacin in patients with coronary-artery disease. N. Engl. J. Med. 1981, 305, 1171–1175. [Google Scholar] [CrossRef]
- Pacold, I.; Hwang, M.H.; Lawless, C.E.; Diamond, P.; Scanlon, P.J.; Loeb, H.S. Effects of indomethacin on coronary hemodynamics, myocardial metabolism and anginal threshold in coronary artery disease. Am. J. Cardiol. 1986, 57, 912–915. [Google Scholar] [CrossRef]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Whittle, B.J. COX-1 and COX-2 products in the gut: Therapeutic impact of COX-2 inhibitors. Gut 2000, 47, 320–325. [Google Scholar] [CrossRef] [Green Version]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Konstam, M.A.; Weir, M.R.; Reicin, A.; Shapiro, D.; Sperling, R.S.; Barr, E.; Gertz, B.J. Cardiovascular thrombotic events in controlled, clinical trials of rofecoxib. Circulation 2001, 104, 2280–2288. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Reilly, I.A.G.; FitzGerald, G.A. Inhibition of Thromboxane Formation In Vivo and Ex Vivo: Implications for Therapy With Platelet Inhibitory Drugs. Blood 1987, 69, 180–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ 2017, 357, j1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viñals, M.; Martínez-González, J.; Badimon, J.J.; Badimon, L. HDL-induced prostacyclin release in smooth muscle cells is dependent on cyclooxygenase-2 (Cox-2). Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3481–3488. [Google Scholar] [CrossRef] [PubMed]
- Catella-Lawson, F.; Reilly, M.P.; Kapoor, S.C.; Cucchiara, A.J.; DeMarco, S.; Tournier, B.; Vyas, S.N.; FitzGerald, G.A. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N. Engl. J. Med. 2001, 345, 1809–1817. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J. CANTOS Trial Group Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- CHMP Withdrawal Assessment Report—Fingolimod. Eur. Med. Agency 2020, 30. Available online: https://www.ema.europa.eu/en/documents/withdrawal-report/withdrawal-assessment-report-canakinumab-novartis_en.pdf (accessed on 13 June 2021).
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Gregan, S.M.; Navin, T.J.; Foxwell, B.M.J.; Davies, A.H.; Feldmann, M. Toll-like receptor-2 mediates inflammation and matrix degradation in human atherosclerosis. Circulation 2009, 120, 2462–2469. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Tuñón, J.; Bäck, M.; Badimón, L.; Bochaton-Piallat, M.-L.; Cariou, B.; Daemen, M.J.; Egido, J.; Evans, P.C.; Francis, S.E.; Ketelhuth, D.F.; et al. Interplay between hypercholesterolaemia and inflammation in atherosclerosis: Translating experimental targets into clinical practice. Eur. J. Prev. Cardiol. 2018, 25, 948–955. [Google Scholar] [CrossRef]
- Micha, R.; Imamura, F.; von Ballmoos, M.W.; Solomon, D.H.; Hernán, M.A.; Ridker, P.M.; Mozaffarian, D. Systematic review and meta-analysis of methotrexate use and risk of cardiovascular disease. Am. J. Cardiol. 2011, 108. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Eikelboom, J.W.; Budgeon, C.A.; Thompson, P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J. Am. Coll. Cardiol. 2013, 61, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.-F.; Ireland, M.A.; Lenderink, T.; et al. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.C.; Quinn, S.; Nasis, A.; Hiew, C.; Roberts-Thomson, P.; Adams, H.; Sriamareswaran, R.; Htun, N.M.; Wilson, W.; Stub, D.; et al. Colchicine in Patients With Acute Coronary Syndrome: The Australian COPS Randomized Clinical Trial. Circulation 2020, 142, 1890–1900. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Steen, D.P.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The SOLID-TIMI 52 randomized clinical trial. JAMA 2014, 312, 1006–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donoghue, M.L.; Glaser, R.; Cavender, M.A.; Aylward, P.E.; Bonaca, M.P.; Budaj, A.; Davies, R.Y.; Dellborg, M.; Fox, K.A.A.; Gutierrez, J.A.T.; et al. Effect of Losmapimod on Cardiovascular Outcomes in Patients Hospitalized With Acute Myocardial Infarction: A Randomized Clinical Trial. JAMA 2016, 315, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Ben-Chetrit, E.; Levy, M. Colchicine: 1998 update. Semin. Arthritis Rheum. 1998, 28, 48–59. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine--Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Fiolet, A.T.L.; Opstal, T.S.J.; Mosterd, A.; Eikelboom, J.W.; Jolly, S.S.; Keech, A.C.; Kelly, P.; Tong, D.C.; Layland, J.; Nidorf, S.M.; et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: A systematic review and meta-analysis of randomized trials. Eur. Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Familial hypercholesterolemia. A genetic regulatory defect in cholesterol metabolism. Am. J. Med. 1975, 58, 147–150. [Google Scholar] [CrossRef]
- Xu, S.; Pelisek, J.; Jin, Z.G. Atherosclerosis Is an Epigenetic Disease. Trends Endocrinol. Metab. 2018, 29, 739–742. [Google Scholar] [CrossRef]
- Kiechl, S.; Lorenz, E.; Reindl, M.; Wiedermann, C.J.; Oberhollenzer, F.; Bonora, E.; Willeit, J.; Schwartz, D.A. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 2002, 347, 185–192. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Study | CANTOS [81] | CIRT [89] | LODOCO1 [90] | LODOCO2 [91] | COLCOT [92] | COPS [93] |
|---|---|---|---|---|---|---|
| Trial population | ||||||
| CV previous disease | Previous MI | Previous MI or multivessel CD | Stable CAD | Stable CAD | MI 30 days before | ACS |
| High CRP mg/L | >2 | No | No | No | No | No |
| Others | DM or metabolic syndrome | Complete percutaneous revascularization | Evidence of CAD | |||
| Trial design | ||||||
| Participating centers | Multicenter | Multicenter | Single-center | Multicenter | Multicenter | Multicenter |
| Design | Double blind | Double blind | Observer blind | Double blind | Double blind | Double blind |
| Study drug and dose | Canakinumab 50;150;300 mg/3 months | Methotrexate 15–20 mg/weekly | Colchicine 0.5 mg/day | Colchicine 0.5 mg/day | Colchicine 0.5 mg/day | Colchicine 0.5 mg/day (twice daily first month) |
| Follow-up (years) | 3.7 | 2.3 | 3 | 2.3 | 1.9 | 1 |
| Sample size | 10,061 | 4786 | 532 | 5522 | 4745 | 795 |
| Characteristics of the participants | ||||||
| Age yr | 61 | 66 | 66.5 | 66 | 60.6 | 59.8 |
| Male % | 72.5 | 81 | 89 | 84.7 | 80.8 | 79.5 |
| DM % | 40 | 68 | 30.5 | 18.2 | 20.2 | 19 |
| Statin % | 93.4 | 85.9 | 95 | 94 | 99 | 98.5 |
| Anti-thrombotic treatment | 95.1 | 86.4 | 93 | 90.2 | 99 | 97.8 |
| Median LDL cholesterol mg/dl | 82.4 | 68 | NR | NR | NR | NR |
| Median CRP mg/L | 4.2 | 1.6 | NR | NR | 4.28 (only 207 patients) | NR |
| End points | ||||||
| Non-fatal MI, stroke or CV death | Positive 150 mg HR 0.85 (0.74–0.98) | Negative HR 1.01 (0.82–1.25) | - | Positive HR 0.69 (0.57–0.83) * | Positive HR 0.77 (0.61–0.96) ** | Negative HR 0.65 (0.38–1.09) *** |
| All-cause mortality | NS | NS | - | NS | NS | HR 8.20 (1.03–65.61) |
| ACS, out-of-hospital cardiac arrest, or non-cardioembolic ischemic stroke | - | - | Positive HR 0.33 (0.18–0.59) | - | - | - |
| NNT (patients) | 156 | - | 11 | 91 | 62 | - |
| Adverse events in active drug | More infection, neutropenia, thrombocytopenia | Higher liver enzyme levels, leukopenia and non-basal cell skin cancers | Intestinal intolerance | Less gout and more myalgia | More gastrointestinal events and pneumonia | NS |
| Cost per year€ | 45,957 | 22.8 | 43.2 | 43.2 | 43.2 | 46.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pello Lázaro, A.M.; Blanco-Colio, L.M.; Franco Peláez, J.A.; Tuñón, J. Anti-Inflammatory Drugs in Patients with Ischemic Heart Disease. J. Clin. Med. 2021, 10, 2835. https://doi.org/10.3390/jcm10132835
Pello Lázaro AM, Blanco-Colio LM, Franco Peláez JA, Tuñón J. Anti-Inflammatory Drugs in Patients with Ischemic Heart Disease. Journal of Clinical Medicine. 2021; 10(13):2835. https://doi.org/10.3390/jcm10132835
Chicago/Turabian StylePello Lázaro, Ana María, Luis M. Blanco-Colio, Juan Antonio Franco Peláez, and José Tuñón. 2021. "Anti-Inflammatory Drugs in Patients with Ischemic Heart Disease" Journal of Clinical Medicine 10, no. 13: 2835. https://doi.org/10.3390/jcm10132835
APA StylePello Lázaro, A. M., Blanco-Colio, L. M., Franco Peláez, J. A., & Tuñón, J. (2021). Anti-Inflammatory Drugs in Patients with Ischemic Heart Disease. Journal of Clinical Medicine, 10(13), 2835. https://doi.org/10.3390/jcm10132835