
The Diagnostic Approach to Mitochondrial Disorders in Children in the Era of Next-Generation Sequencing: A 4-Year Cohort Study


 ,
,  ,
,  , ,
, ,  ,
,  , , and add
Show full author list
, , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Data
2.2. Histochemical and Spectrophotometric Investigations in Muscle Biopsy
2.3. Molecular Studies
2.4. Statistical Analyses
3. Results
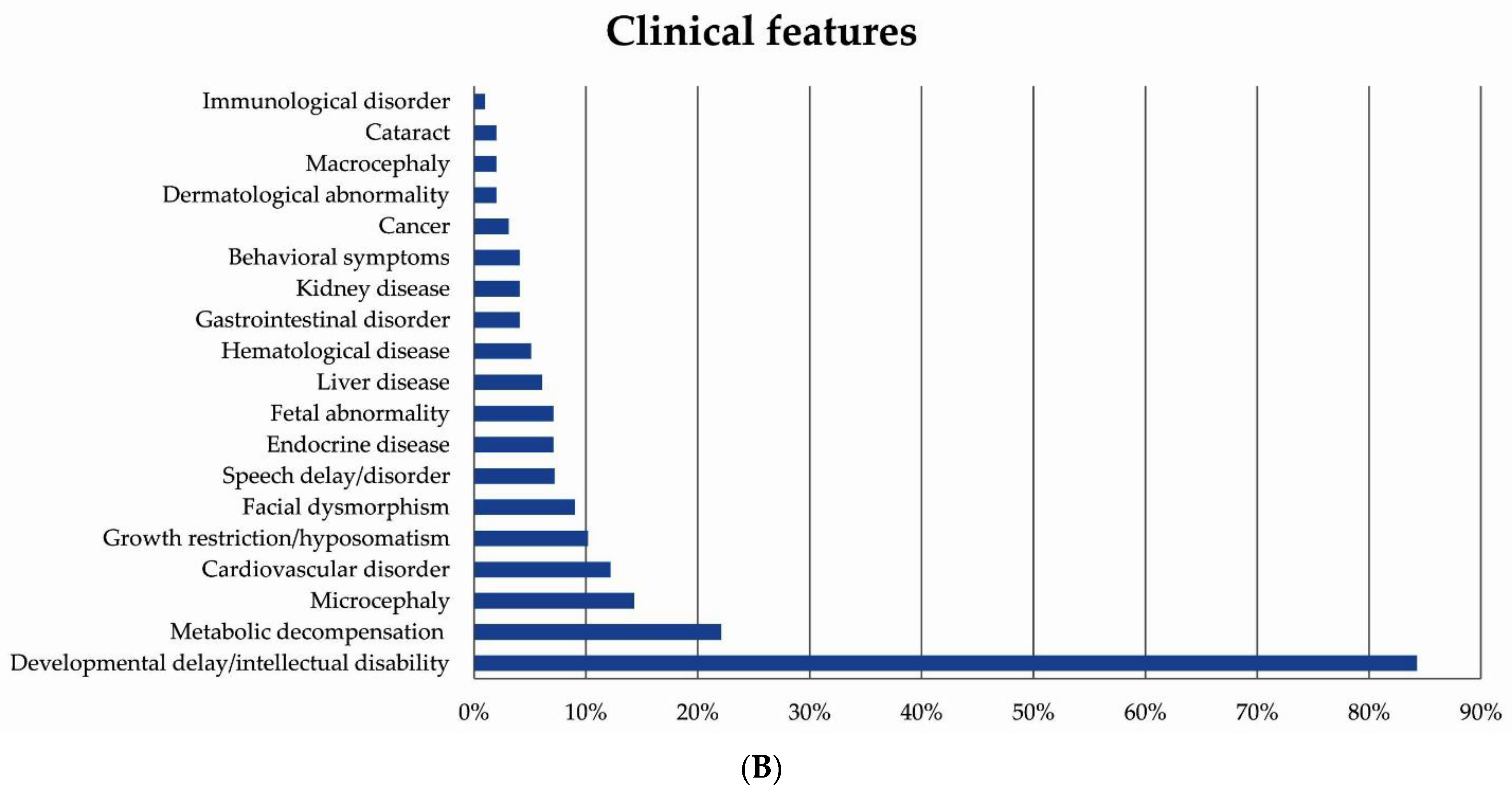
3.1. Patient Demographics and Clinical Presentations
3.2. Neuroimaging Features
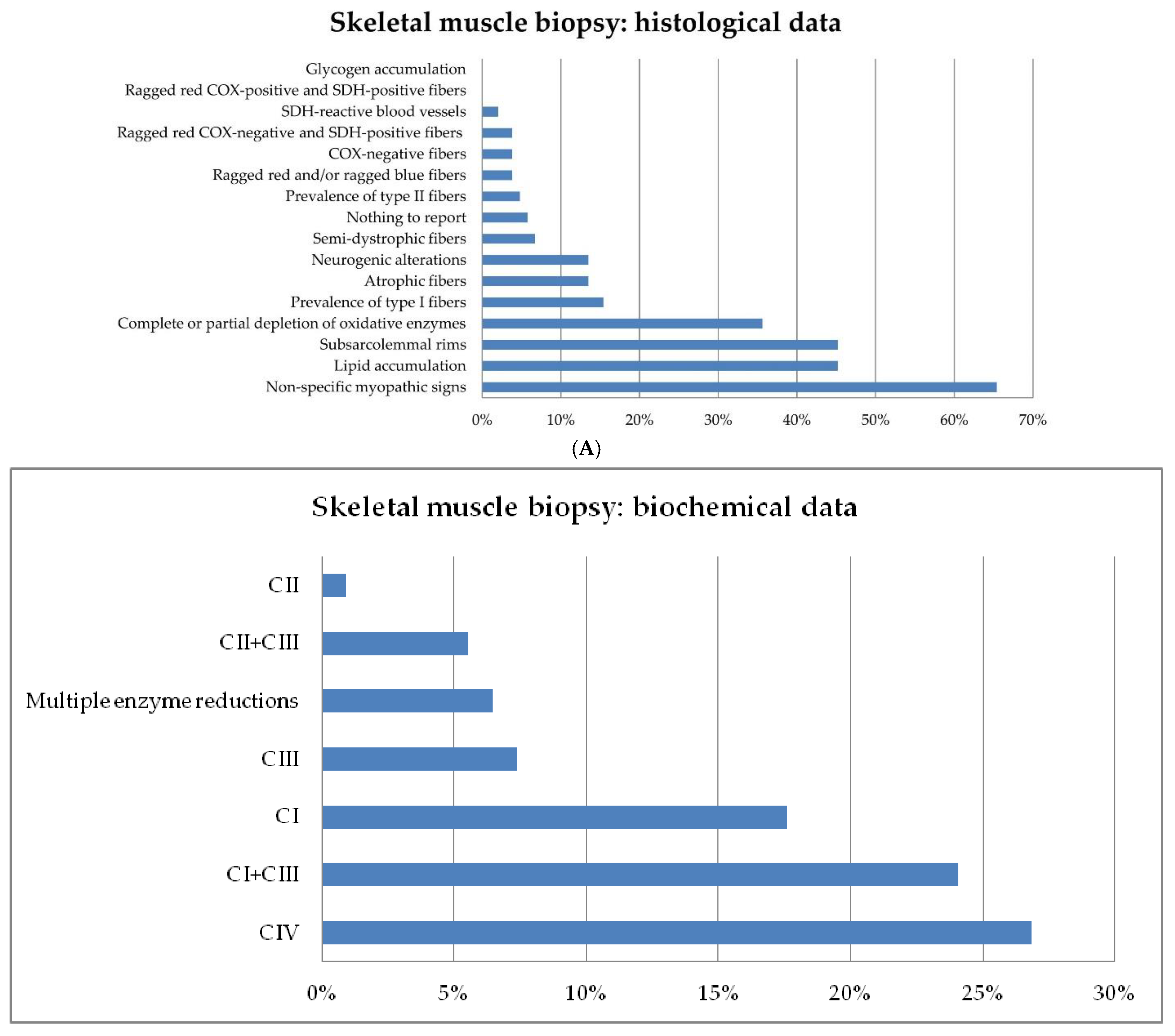
3.3. Skeletal Muscle Biopsy: Histological and Biochemical Analyses
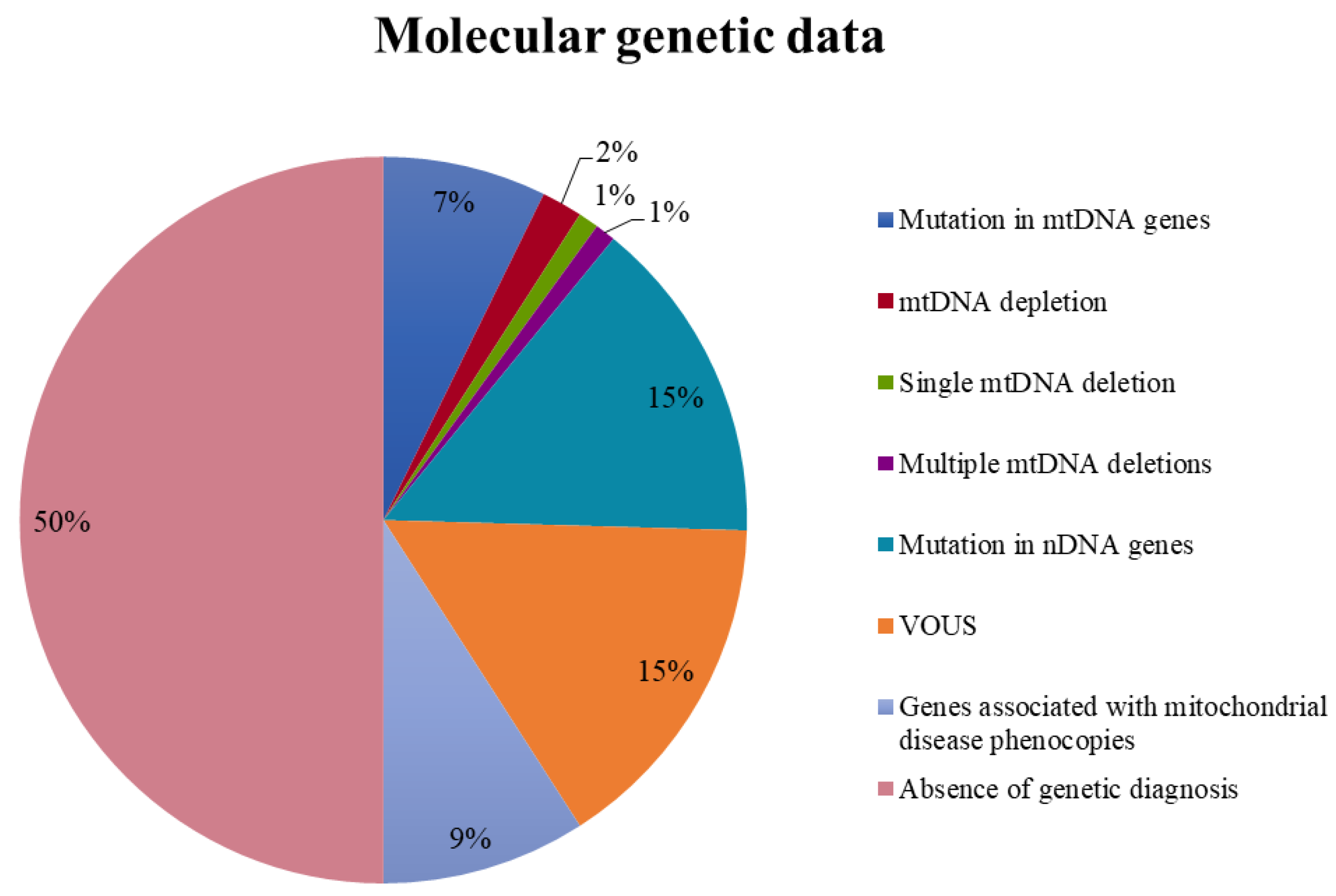
3.4. Molecular Genetics
3.5. Predicting a Molecular Diagnosis of MD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| COX = cytochrome c oxidase; SDH = succinate dehydrogenase; NADH = nicotinamide adenine dinucleotide; CI = complex I; CII= complex II; CIII = complex III; CS = citrate synthase; n.a. = data not available. DD/ID = developmental delay/intellectual disability; WM = white matter; BG =basal ganglia; MRS = magnetic resonance spectroscopy; RRF: ragged red fibers. |
References
- Munnich, A.; Rustin, P. Clinical spectrum and diagnosis of mitochondrial disorders. Am. J. Med. Genet. 2001, 106, 4–17. [Google Scholar] [CrossRef]
- Debray, F.G.; Lambert, M.; Mitchell, G.A. Disorders of mitochondrial function. Curr. Opin. Pediatr. 2008, 20, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef]
- Tan, J.; Wagner, M.; Stenton, S.L.; Strom, T.M.; Wortmann, S.B.; Prokisch, H.; Meitinger, T.; Oexle, K.; Klopstock, T. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine 2020, 54, 102730. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Lim, A.; Gorman, G. Epidemiology of Mitochondrial Disease. In Diagnosis and Management of Mitochondrial Disorders; Springer International Publishing: Cham, Switzerland, 2019; pp. 63–79. [Google Scholar]
- Mancuso, M.; Klopstock, T. Diagnosis and Management of Mitochondrial Disorders; Springer: Berlin/Heidelberg, Germany, 2019; ISBN 9783030055172. [Google Scholar]
- DiMauro, S. Mitochondrial diseases. Biochim. Biophys. Acta Bioenergy 2004, 1658, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torraco, A.; Diaz, F.; Vempati, U.D.; Moraes, C.T. Mouse models of oxidative phosphorylation defects: Powerful tools to study the pathobiology of mitochondrial diseases. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafai, S.B.; Mootha, V.K. A common pathway for a rare disease? Science 2013, 342, 1453–1454. [Google Scholar] [CrossRef]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef]
- Moslemi, A.R.; Darin, N. Molecular genetic and clinical aspects of mitochondrial disorders in childhood. Mitochondrion 2007, 7, 241–252. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Mayr, J.A.; Nuoffer, J.M.; Prokisch, H.; Sperl, W. A Guideline for the Diagnosis of Pediatric Mitochondrial Disease: The Value of Muscle and Skin Biopsies in the Genetics Era. Neuropediatrics 2017, 48, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Muraresku, C.C.; McCormick, E.M.; Falk, M.J. Mitochondrial Disease: Advances in Clinical Diagnosis, Management, Therapeutic Development, and Preventative Strategies. Curr. Genet. Med. Rep. 2018, 6, 62–72. [Google Scholar] [CrossRef]
- Bricout, M.; Grévent, D.; Lebre, A.S.; Rio, M.; Desguerre, I.; de Lonlay, P.; Valayannopoulos, V.; Brunelle, F.; Rötig, A.; Munnich, A.; et al. Brain imaging in mitochondrial respiratory chain deficiency: Combination of brain MRI features as a useful tool for genotype/phenotype correlations. J. Med. Genet. 2014, 51, 429–435. [Google Scholar] [CrossRef]
- Parikh, S.; Karaa, A.; Goldstein, A.; Bertini, E.S.; Chinnery, P.F.; Christodoulou, J.; Cohen, B.H.; Davis, R.L.; Falk, M.J.; Fratter, C.; et al. Diagnosis of possible’ mitochondrial disease: An existential crisis. J. Med. Genet. 2019, 56, 123–130. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease in children. J. Intern. Med. 2020, 87, 609–633. [Google Scholar] [CrossRef]
- Nascimento, A.; Ortez, C.; Jou, C.; O’Callaghan, M.; Ramos, F.; Garcia-Cazorla, À. Neuromuscular Manifestations in Mitochondrial Diseases in Children. Semin. Pediatr. Neurol. 2016, 23, 290–305. [Google Scholar] [CrossRef]
- Witters, P.; Saada, A.; Honzik, T.; Tesarova, M.; Kleinle, S.; Horvath, R.; Goldstein, A.; Morava, E. Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genet. Med. 2018, 20, 444–451. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, J.M.; Tarnopolsky, M.A. Pathology of skeletal muscle in mitochondrial disorders. Mitochondrion 2004, 4, 441–452. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Ermani, M.; Salviati, L.; Angelini, C. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion 2011, 11, 893–904. [Google Scholar] [CrossRef]
- He, L.; Chinnery, P.F.; Durham, S.E.; Blakely, E.L.; Wardell, T.M.; Borthwick, G.M.; Taylor, R.W.; Turnbull, D.M. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002, 30, e68. [Google Scholar] [CrossRef]
- Bai, R.K.; Wong, L.J. Simultaneous detection and quantification of mitochondrial DNA deletion(s), depletion, and over-replication in patients with mitochondrial disease. J. Mol. Diagn. 2005, 7, 613–622. [Google Scholar] [CrossRef] [Green Version]
- de Michele, G.; Sorrentino, P.; Nesti, C.; Rubegni, A.; Ruggiero, L.; Peluso, S.; Antenora, A.; Quarantelli, M.; Filla, A.; de Michele, G.; et al. Reversible valproate-induced subacute encephalopathy associated with a MT-ATP8 variant in the mitochondrial genome. Front. Neurol. 2018, 9, 728. [Google Scholar] [CrossRef]
- Craven, L.; Alston, C.L.; Taylor, R.W.; Turnbull, D.M. Recent Advances in Mitochondrial Disease. Annu. Rev. Genom. Hum. Genet. 2017, 18, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Haack, T.B.; Haberberger, B.; Frisch, E.M.; Wieland, T.; Iuso, A.; Gorza, M.; Strecker, V.; Graf, E.; Mayr, J.A.; Herberg, U.; et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J. Med. Genet. 2012, 49, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Neveling, K.; Feenstra, I.; Gilissen, C.; Hoefsloot, L.H.; Kamsteeg, E.J.; Mensenkamp, A.R.; Rodenburg, R.J.T.; Yntema, H.G.; Spruijt, L.; Vermeer, S.; et al. A Post-Hoc Comparison of the Utility of Sanger Sequencing and Exome Sequencing for the Diagnosis of Heterogeneous Diseases. Hum. Mutat. 2013, 34, 1721–1726. [Google Scholar] [CrossRef]
- Wortmann, S.B.; Koolen, D.A.; Smeitink, J.A.; van den Heuvel, L.; Rodenburg, R.J. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J. Inherit. Metab. Dis. 2015, 38, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Koenig, M.K. Presentation and Diagnosis of Mitochondrial Disorders in Children. Pediatr. Neurol. 2008, 38, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Kisler, J.E.; Whittaker, R.G.; Mcfarland, R. Mitochondrial diseases in childhood: A clinical approach to investigation and management. Dev. Med. Child Neurol. 2010, 52, 422–433. [Google Scholar] [CrossRef]
- El Sabbagh, S.; Lebre, A.S.; Bahi-Buisson, N.; Delonlay, P.; Soufflet, C.; Boddaert, N.; Rio, M.; Rötig, A.; Dulac, O.; Munnich, A.; et al. Epileptic phenotypes in children with respiratory chain disorders. Epilepsia 2010, 51, 1225–1235. [Google Scholar] [CrossRef]
- Verity, C.M.; Winstone, A.M.; Stellitano, L.; Krishnakumar, D.; Will, R.; Mcfarland, R. The clinical presentation of mitochondrial diseases in children with progressive intellectual and neurological deterioration: A national, prospective, population-based study. Dev. Med. Child Neurol. 2010, 52, 434–440. [Google Scholar] [CrossRef]
- Chevallier, J.A.; Von Allmen, G.K.; Koenig, M.K. Seizure semiology and EEG findings in mitochondrial diseases. Epilepsia 2014, 55, 707–712. [Google Scholar] [CrossRef]
- Ticci, C.; Sicca, F.; Ardissone, A.; Bertini, E.; Carelli, V.; Diodato, D.; Di Vito, L.; Filosto, M.; La Morgia, C.; Lamperti, C.; et al. Mitochondrial epilepsy: A cross-sectional nationwide Italian survey. Neurogenetics 2020, 21, 87–96. [Google Scholar] [CrossRef]
- Hegde, A.N.; Mohan, S.; Lath, N.; Lim, C.C.T. Differential diagnosis for bilateral abnormalities of the basal ganglia and thalamus. Radiographics 2011, 31, 5–30. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.V.; Hoon, J. Possible mechanisms in infants for selective basal ganglia damage from asphyxia, kernicterus, or mitochondrial encephalopathies. J. Child Neurol. 2000, 15, 588–591. [Google Scholar] [CrossRef] [PubMed]
- de Beaurepaire, I.; Grévent, D.; Rio, M.; Desguerre, I.; de Lonlay, P.; Levy, R.; Dangouloff-Ros, V.; Bonnefont, J.P.; Barcia, G.; Funalot, B.; et al. High predictive value of brain MRI imaging in primary mitochondrial respiratory chain deficiency. J. Med. Genet. 2018, 55, 378–383. [Google Scholar] [CrossRef]
- Koga, Y.; Koga, A.; Iwanaga, R.; Akita, Y.; Tubone, J.; Matsuishi, T.; Takane, N.; Sato, Y.; Kato, H. Single-fiber analysis of mitochondrial A3243G mutation in four different phenotypes. Acta Neuropathol. 2000, 99, 186–190. [Google Scholar] [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Lamont, P.J.; Surtees, R.; Woodward, C.E.; Leonard, J.V.; Wood, N.W.; Harding, A.E. Clinical and laboratory findings in referrals for mitochondrial DNA analysis. Arch. Dis. Child. 1998, 79, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New perspective in diagnostics of mitochondrial disorders: Two years’ experience with whole-exome sequencing at a national paediatric centre. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef] [Green Version]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Clark, M.M.; Hildreth, A.; Batalov, S.; Ding, Y.; Chowdhury, S.; Watkins, K.; Ellsworth, K.; Camp, B.; Kint, C.I.; Yacoubian, C.; et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 2019, 11, eaat6177. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolomeo, D.; Orsucci, D.; Nesti, C.; Baldacci, J.; Battini, R.; Bruno, C.; Bruno, G.; Cassandrini, D.; Doccini, S.; Donati, M.A.; et al. The Diagnostic Approach to Mitochondrial Disorders in Children in the Era of Next-Generation Sequencing: A 4-Year Cohort Study. J. Clin. Med. 2021, 10, 3222. https://doi.org/10.3390/jcm10153222
Tolomeo D, Orsucci D, Nesti C, Baldacci J, Battini R, Bruno C, Bruno G, Cassandrini D, Doccini S, Donati MA, et al. The Diagnostic Approach to Mitochondrial Disorders in Children in the Era of Next-Generation Sequencing: A 4-Year Cohort Study. Journal of Clinical Medicine. 2021; 10(15):3222. https://doi.org/10.3390/jcm10153222
Chicago/Turabian StyleTolomeo, Deborah, Daniele Orsucci, Claudia Nesti, Jacopo Baldacci, Roberta Battini, Claudio Bruno, Giorgia Bruno, Denise Cassandrini, Stefano Doccini, M. Alice Donati, and et al. 2021. "The Diagnostic Approach to Mitochondrial Disorders in Children in the Era of Next-Generation Sequencing: A 4-Year Cohort Study" Journal of Clinical Medicine 10, no. 15: 3222. https://doi.org/10.3390/jcm10153222
APA StyleTolomeo, D., Orsucci, D., Nesti, C., Baldacci, J., Battini, R., Bruno, C., Bruno, G., Cassandrini, D., Doccini, S., Donati, M. A., Ferrari, A., Fiori, S., Fiorillo, C., Guerrini, R., Mari, F., Montomoli, M., Pochiero, F., Procopio, E., Ruggiero, L., ... Santorelli, F. M. (2021). The Diagnostic Approach to Mitochondrial Disorders in Children in the Era of Next-Generation Sequencing: A 4-Year Cohort Study. Journal of Clinical Medicine, 10(15), 3222. https://doi.org/10.3390/jcm10153222