
Still’s Disease in the Constellation of Hyperinflammatory Syndromes: A Link with Kawasaki Disease?


Abstract
:1. Conceptual Evolution of Still’s Disease
2. Conceptual Evolution of Kawasaki Disease
3. Still’s Disease and KD, How Much Are They Related?
Is It Possible to Link Them Clinically?
4. How Their Pathophysiology Could Be Similar?
Innate Immune System Activation
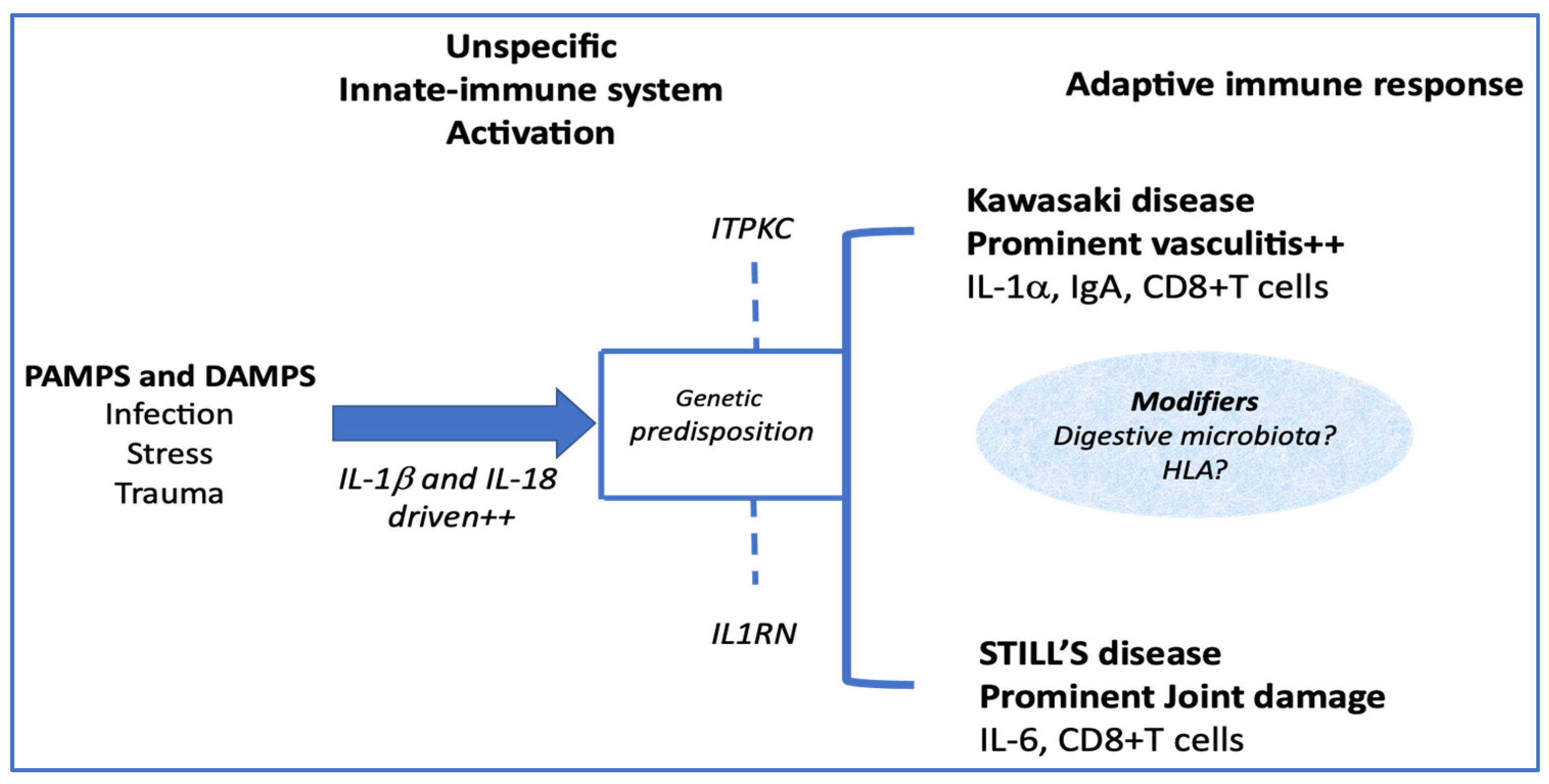
5. How Much Are They Different?
5.1. Clinical Differences
5.2. Adaptive Immune System Induction
5.3. Endothelial Activation
6. Conclusions
7. Take Home Messages
7.1. What Are the Key Elements That Our Analysis Points Out?
- –
- KD and Still’s disease share a number of clinical and biological similarities but essentially differ from their respective evolutions.
- –
- Both diseases are underpinned by related physiopathological mechanisms encompassing a potent activation of innate immunity.
- –
- Rather, they should be considered as belonging to a common clinical spectrum, the extremes of which depend on a variable influence of adaptive immunity.
- –
- The preferential endothelial activation of KD compared to Still’s disease is strongly enhanced by the release of interleukin 1 alpha and the direct toxicity of the IgA toward the endothelial cells.
7.2. How Might This Impact on Clinical Practice or Future Developments?
- –
- The deep link between these two syndromes (rather than diseases) implies considering them together to resolve physiopathological or therapeutic research questions.
- –
- The crucial role of interleukin 1 alpha in inducing cardiac inflammation of KD must be taken into account when choosing targeted biotherapies.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Still, G.F. On a Form of Chronic Joint Disease in Children. Med. Chir. Trans. 1897, 80, 47–60.9. [Google Scholar] [CrossRef]
- Woo, P.; Wulffraat, N.M.; Tyndall, A. Barbara Ansell: A Person Ahead of Her Time. Ann. Rheum. Dis. 2019, 78, 725–728. [Google Scholar] [CrossRef]
- Kaiser, H.; Mayer, S. Diamantberger (1864–1944). The first person to describe juvenile chronic arthritis. Z. Rheumatol. 2009, 68, 264–266. [Google Scholar] [CrossRef]
- Hadchouel, M.; Prieur, A.M.; Griscelli, C. Acute Hemorrhagic, Hepatic, and Neurologic Manifestations in Juvenile Rheumatoid Arthritis: Possible Relationship to Drugs or Infection. J. Pediatr. 1985, 106, 561–566. [Google Scholar] [CrossRef]
- Quartier, P.; Taupin, P.; Bourdeaut, F.; Lemelle, I.; Pillet, P.; Bost, M.; Sibilia, J.; Koné-Paut, I.; Gandon-Laloum, S.; LeBideau, M.; et al. Efficacy of Etanercept for the Treatment of Juvenile Idiopathic Arthritis According to the Onset Type. Arthritis Rheumatol. 2003, 48, 1093–1101. [Google Scholar] [CrossRef]
- Pignatti, P.; Vivarelli, M.; Meazza, C.; Rizzolo, M.G.; Martini, A.; De Benedetti, F. Abnormal Regulation of Interleukin 6 in Systemic Juvenile Idiopathic Arthritis. J. Rheumatol. 2001, 28, 1670–1676. [Google Scholar]
- Prieur, A.M.; Roux-Lombard, P.; Dayer, J.M. Dynamics of Fever and the Cytokine Network in Systemic Juvenile Arthritis. Rev. Rhum. Engl. Ed. 1996, 63, 163–170. [Google Scholar] [PubMed]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of Interleukin-1 (IL-1) in the Pathogenesis of Systemic Onset Juvenile Idiopathic Arthritis and Clinical Response to IL-1 Blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef]
- McDermott, M.F.; Aksentijevich, I.; Galon, J.; McDermott, E.M.; Ogunkolade, B.W.; Centola, M.; Mansfield, E.; Gadina, M.; Karenko, L.; Pettersson, T.; et al. Germline Mutations in the Extracellular Domains of the 55 KDa TNF Receptor, TNFR1, Define a Family of Dominantly Inherited Autoinflammatory Syndromes. Cell 1999, 97, 133–144. [Google Scholar] [CrossRef]
- Tartey, S.; Kanneganti, T.-D. Inflammasomes in the Pathophysiology of Autoinflammatory Syndromes. J. Leukoc. Biol. 2020, 107, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T. Acute febrile mucocutaneous syndrome with lymphoid involvement with specific desquamation of the fingers and toes in children. Arerugi Allergy 1967, 16, 178–222. [Google Scholar] [PubMed]
- Kato, H.; Koike, S.; Yokoyama, T. Kawasaki Disease: Effect of Treatment on Coronary Artery Involvement. Pediatrics 1979, 63, 175–179. [Google Scholar]
- McCrindle, B.W.; Rowley, A.H.; Newburger, J.W.; Burns, J.C.; Bolger, A.F.; Gewitz, M.; Baker, A.L.; Jackson, M.A.; Takahashi, M.; Shah, P.B.; et al. Diagnosis, Treatment, and Long-Term Management of Kawasaki Disease: A Scientific Statement for Health Professionals from the American Heart Association. Circulation 2017, 135, e927–e999. [Google Scholar] [CrossRef] [PubMed]
- Kessel, C.; Lippitz, K.; Weinhage, T.; Hinze, C.; Wittkowski, H.; Holzinger, D.; Fall, N.; Grom, A.A.; Gruen, N.; Foell, D. Proinflammatory Cytokine Environments Can Drive Interleukin-17 Overexpression by γ/δ T Cells in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2017, 69, 1480–1494. [Google Scholar] [CrossRef]
- Lee, Y.; Wakita, D.; Dagvadorj, J.; Shimada, K.; Chen, S.; Huang, G.; Lehman, T.J.A.; Fishbein, M.C.; Hoffman, H.M.; Crother, T.R.; et al. IL-1 Signaling Is Critically Required in Stromal Cells in Kawasaki Disease Vasculitis Mouse Model: Role of Both IL-1α and IL-1β. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2605–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koné-Paut, I.; Tellier, S.; Belot, A.; Brochard, K.; Guitton, C.; Marie, I.; Meinzer, U.; Cherqaoui, B.; Galeotti, C.; Boukhedouni, N.; et al. Phase II Open Label Study of Anakinra in Intravenous Immunoglobulin-Resistant Kawasaki Disease. Arthritis Rheumatol. 2021, 73, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Martini, A. Juvenile Idiopathic Arthritis. Lancet 2007, 369, 767–778. [Google Scholar] [CrossRef] [Green Version]
- Makino, N.; Nakamura, Y.; Yashiro, M.; Kosami, K.; Matsubara, Y.; Ae, R.; Aoyama, Y.; Yanagawa, H. Nationwide Epidemiologic Survey of Kawasaki Disease in Japan, 2015–2016. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2019, 61, 397–403. [Google Scholar] [CrossRef]
- Robinson, C.; Chanchlani, R.; Gayowsky, A.; Brar, S.; Darling, E.; Demers, C.; Klowak, J.; Knight, B.; Kuenzig, E.; Mondal, T.; et al. Incidence and Short-Term Outcomes of Kawasaki Disease. Pediatr. Res. 2021. [Google Scholar] [CrossRef]
- Salo, E.; Griffiths, E.P.; Farstad, T.; Schiller, B.; Nakamura, Y.; Yashiro, M.; Uehara, R.; Best, B.M.; Burns, J.C. Incidence of Kawasaki Disease in Northern European Countries. Pediatr. Int. Off. J. Jpn. Pediatr. Soc. 2012, 54, 770–772. [Google Scholar] [CrossRef]
- Stojanov, S.; Lapidus, S.; Chitkara, P.; Feder, H.; Salazar, J.C.; Fleisher, T.A.; Brown, M.R.; Edwards, K.M.; Ward, M.M.; Colbert, R.A.; et al. Periodic Fever, Aphthous Stomatitis, Pharyngitis, and Adenitis (PFAPA) Is a Disorder of Innate Immunity and Th1 Activation Responsive to IL-1 Blockade. Proc. Natl. Acad. Sci. USA 2011, 108, 7148–7153. [Google Scholar] [CrossRef] [Green Version]
- Berntson, L.; Andersson Gäre, B.; Fasth, A.; Herlin, T.; Kristinsson, J.; Lahdenne, P.; Marhaug, G.; Nielsen, S.; Pelkonen, P.; Rygg, M.; et al. Incidence of Juvenile Idiopathic Arthritis in the Nordic Countries. A Population Based Study with Special Reference to the Validity of the ILAR and EULAR Criteria. J. Rheumatol. 2003, 30, 2275–2282. [Google Scholar]
- Piram, M.; Darce Bello, M.; Tellier, S.; Di Filippo, S.; Boralevi, F.; Madhi, F.; Meinzer, U.; Cimaz, R.; Piedvache, C.; Koné-Paut, I. Defining the Risk of First Intravenous Immunoglobulin Unresponsiveness in Non-Asian Patients with Kawasaki Disease. Sci. Rep. 2020, 10, 3125. [Google Scholar] [CrossRef] [Green Version]
- Bruck, N.; Schnabel, A.; Hedrich, C.M. Current Understanding of the Pathophysiology of Systemic Juvenile Idiopathic Arthritis (SJIA) and Target-Directed Therapeutic Approaches. Clin. Immunol. 2015, 159, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Lefèvre-Utile, A.; Galeotti, C.; Koné-Paut, I. Coronary Artery Abnormalities in Children with Systemic-Onset Juvenile Idiopathic Arthritis. Jt. Bone Spine 2014, 81, 257–259. [Google Scholar] [CrossRef]
- Put, K.; Avau, A.; Brisse, E.; Mitera, T.; Put, S.; Proost, P.; Bader-Meunier, B.; Westhovens, R.; Van den Eynde, B.J.; Orabona, C.; et al. Cytokines in Systemic Juvenile Idiopathic Arthritis and Haemophagocytic Lymphohistiocytosis: Tipping the Balance between Interleukin-18 and Interferon-γ. Rheumatol. Oxf. Engl. 2015, 54, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracaglia, C.; de Graaf, K.; Pires Marafon, D.; Guilhot, F.; Ferlin, W.; Prencipe, G.; Caiello, I.; Davì, S.; Schulert, G.; Ravelli, A.; et al. Elevated Circulating Levels of Interferon-γ and Interferon-γ-Induced Chemokines Characterise Patients with Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis. Ann. Rheum. Dis. 2017, 76, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gong, F.; Zhu, W.; Fu, S.; Zhang, Q. Macrophage Activation Syndrome in Kawasaki Disease: More Common than We Thought? Semin. Arthritis Rheum. 2015, 44, 405–410. [Google Scholar] [CrossRef]
- Wittkowski, H.; Hirono, K.; Ichida, F.; Vogl, T.; Ye, F.; Yanlin, X.; Saito, K.; Uese, K.; Miyawaki, T.; Viemann, D.; et al. Acute Kawasaki Disease Is Associated with Reverse Regulation of Soluble Receptor for Advance Glycation End Products and Its Proinflammatory Ligand S100A12. Arthritis Rheum. 2007, 56, 4174–4181. [Google Scholar] [CrossRef]
- Wang, Y.; Qian, S.Y.; Yuan, Y.; Wang, Q.; Gao, L.; Chen, X.; Yu, X.; Zhen, Z. Do Cytokines Correlate with Refractory Kawasaki Disease in Children? Clin. Chim. Acta Int. J. Clin. Chem. 2020, 506, 222–227. [Google Scholar] [CrossRef]
- Jia, C.; Zhang, J.; Chen, H.; Zhuge, Y.; Chen, H.; Qian, F.; Zhou, K.; Niu, C.; Wang, F.; Qiu, H.; et al. Endothelial Cell Pyroptosis Plays an Important Role in Kawasaki Disease via HMGB1/RAGE/Cathespin B Signaling Pathway and NLRP3 Inflammasome Activation. Cell Death Dis. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Armaroli, G.; Verweyen, E.; Pretzer, C.; Kessel, K.; Hirono, K.; Ichida, F.; Okabe, M.; Cabral, D.A.; Foell, D.; Brown, K.L.; et al. Monocyte-Derived Interleukin-1β As the Driver of S100A12-Induced Sterile Inflammatory Activation of Human Coronary Artery Endothelial Cells: Implications for the Pathogenesis of Kawasaki Disease. Arthritis Rheumatol. 2019, 71, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Alphonse, M.P.; Duong, T.T.; Shumitzu, C.; Hoang, T.L.; McCrindle, B.W.; Franco, A.; Schurmans, S.; Philpott, D.J.; Hibberd, M.L.; Burns, J.; et al. Inositol-Triphosphate 3-Kinase C Mediates Inflammasome Activation and Treatment Response in Kawasaki Disease. J. Immunol. 2016, 197, 3481–3489. [Google Scholar] [CrossRef] [Green Version]
- Arthur, V.L.; Shuldiner, E.; Remmers, E.F.; Hinks, A.; Grom, A.A.; Foell, D.; Martini, A.; Gattorno, M.; Özen, S.; Prahalad, S.; et al. IL1RN Variation Influences Both Disease Susceptibility and Response to Recombinant Human Interleukin-1 Receptor Antagonist Therapy in Systemic Juvenile Idiopathic Arthritis. Arthritis Rheumatol. 2018, 70, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noval Rivas, M.; Wakita, D.; Franklin, M.K.; Carvalho, T.T.; Abolhesn, A.; Gomez, A.C.; Fishbein, M.C.; Chen, S.; Lehman, T.J.; Sato, K.; et al. Intestinal Permeability and IgA Provoke Immune Vasculitis Linked to Cardiovascular Inflammation. Immunity 2019, 51, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Hamamichi, Y.; Ichida, F.; Yu, X.; Hirono, K.I.; Uese, K.I.; Hashimoto, I.; Tsubata, S.; Yoshida, T.; Futatani, T.; Kanegane, H.; et al. Neutrophils and Mononuclear Cells Express Vascular Endothelial Growth Factor in Acute Kawasaki Disease: Its Possible Role in Progression of Coronary Artery Lesions. Pediatr. Res. 2001, 49, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Breunis, W.B.; Davila, S.; Shimizu, C.; Oharaseki, T.; Takahashi, K.; van Houdt, M.; Khor, C.C.; Wright, V.J.; Levin, M.; Burns, J.C.; et al. Disruption of Vascular Homeostasis in Patients with Kawasaki Disease: Involvement of Vascular Endothelial Growth Factor and Angiopoietins. Arthritis Rheum. 2012, 64, 306–315. [Google Scholar] [CrossRef]
- Malik, A.; Kanneganti, T.-D. Function and Regulation of IL-1α in Inflammatory Diseases and Cancer. Immunol. Rev. 2018, 281, 124–137. [Google Scholar] [CrossRef]
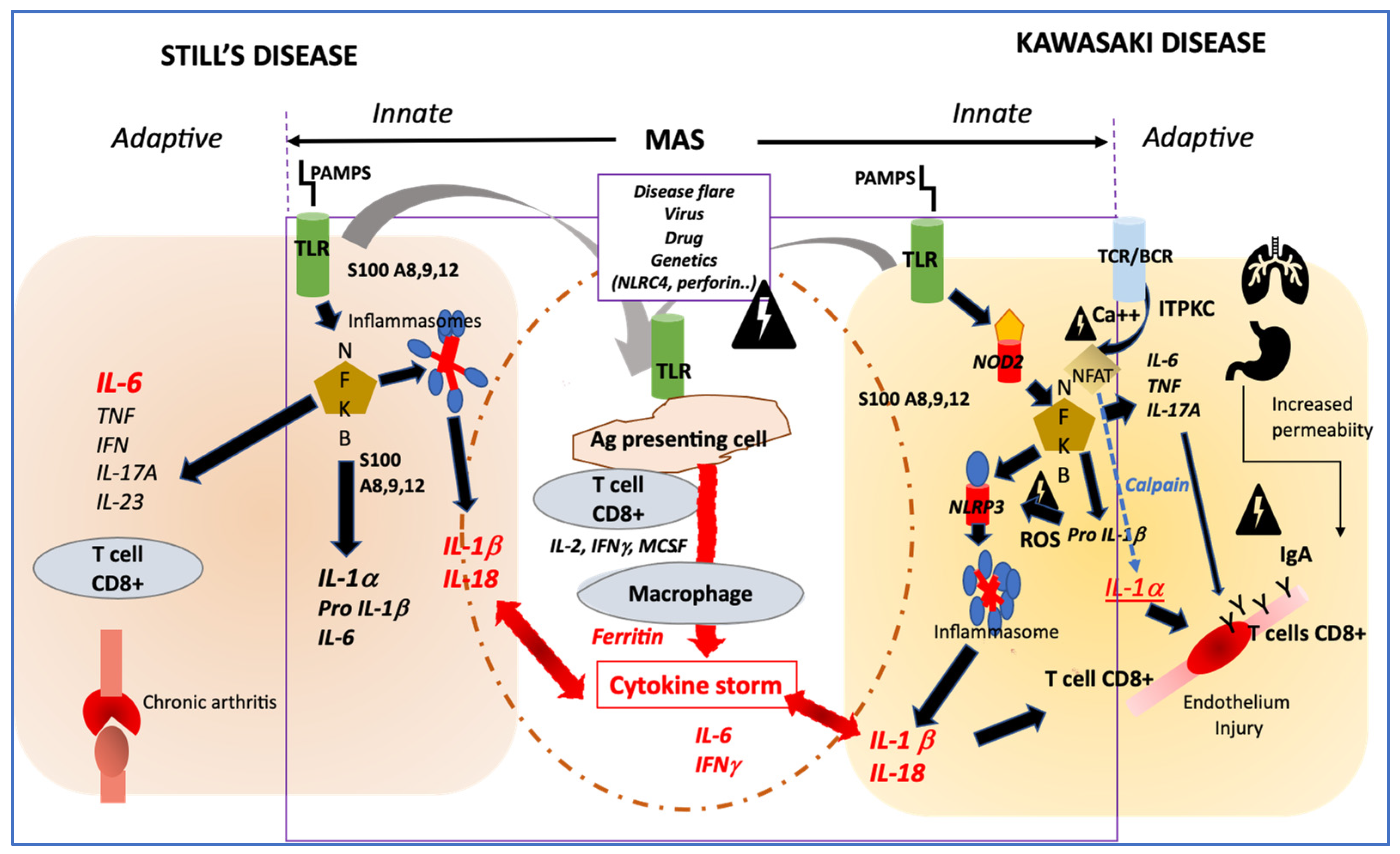

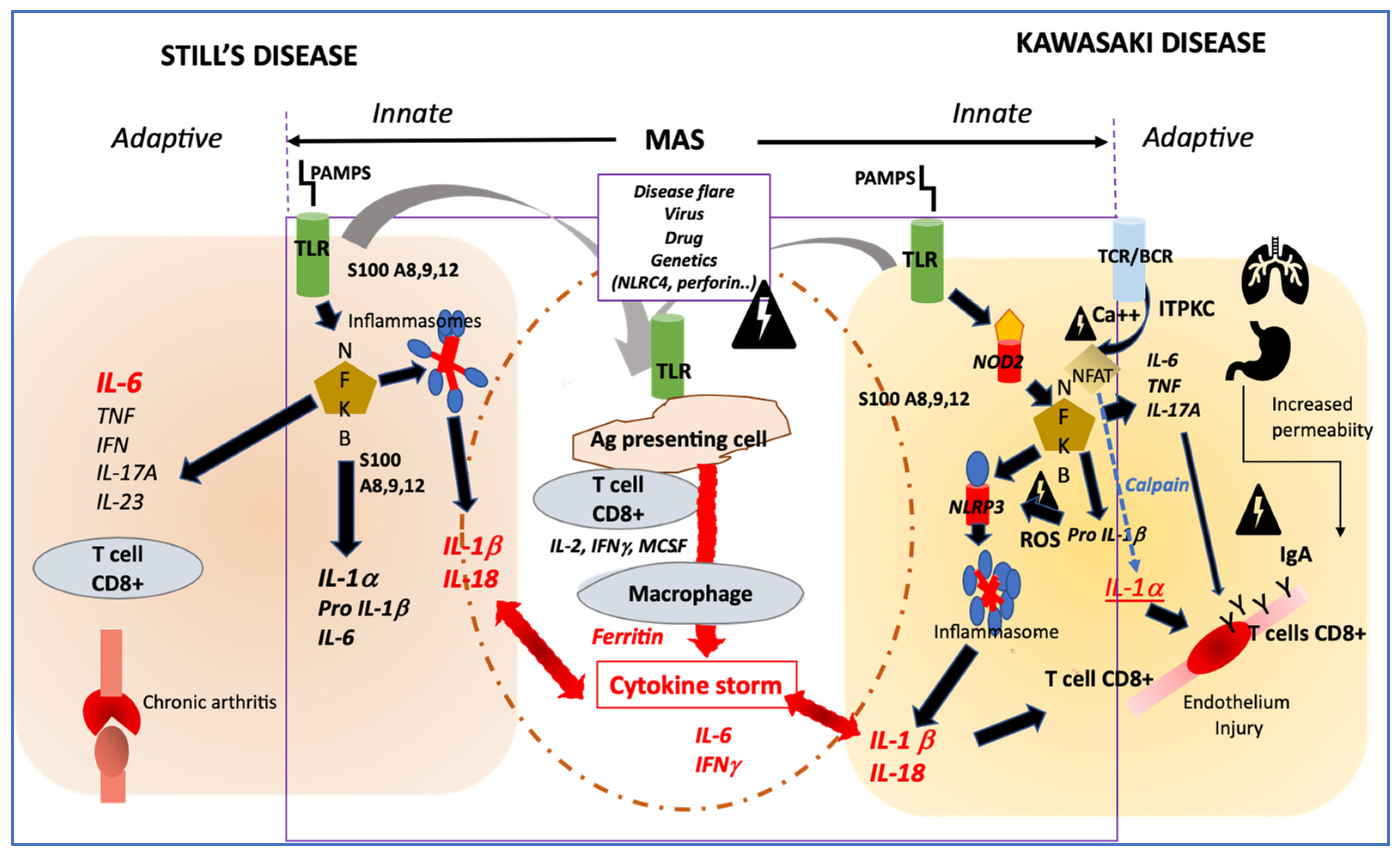{kind=link}
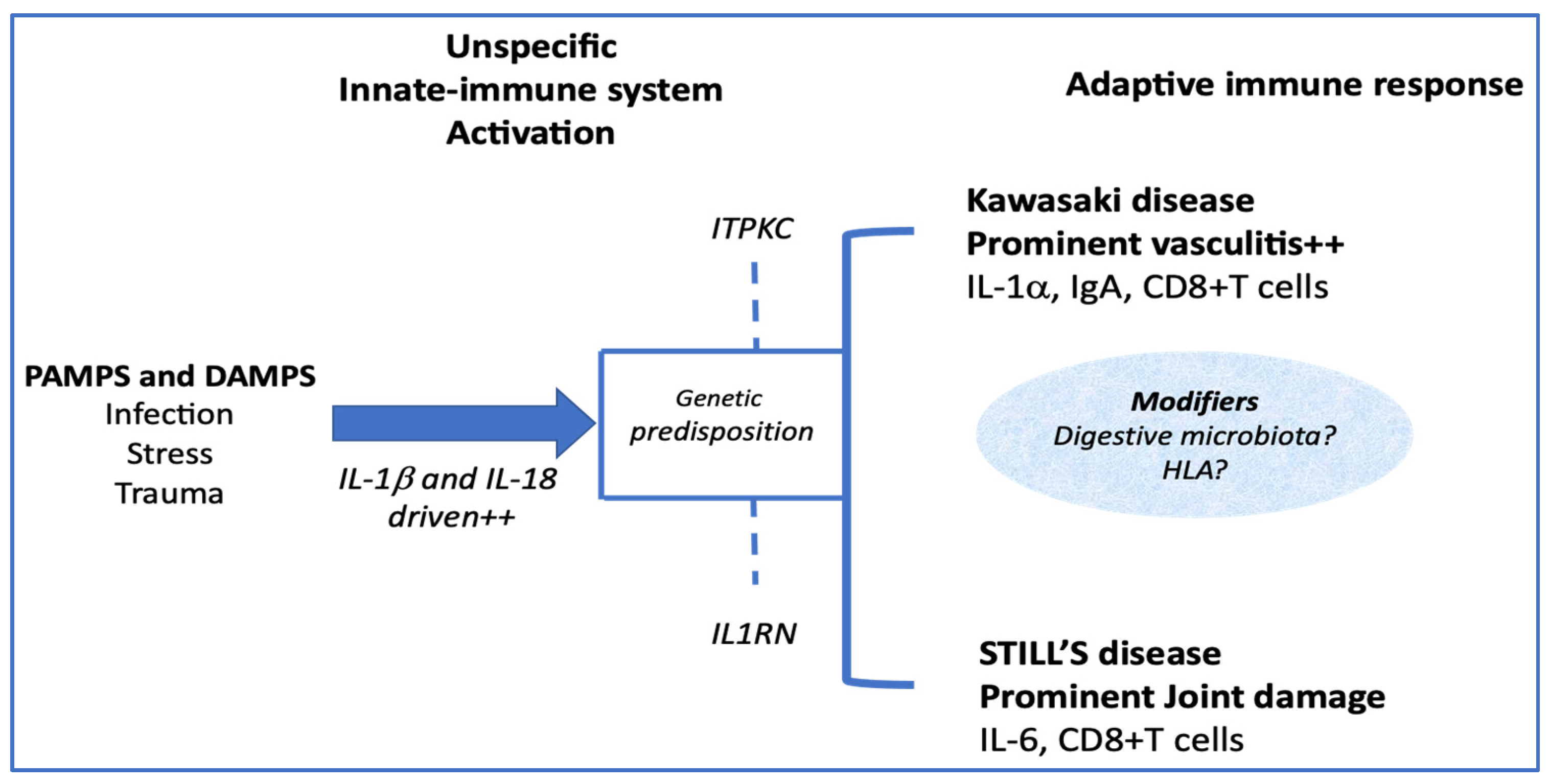{kind=link}
| Clinical Sign | Still’s Disease | Kawasaki Disease |
|---|---|---|
| Estimated incidence | Children 0.6/<16 years [22] All ethnicities, minor prevalence and incidence variations | Japan: 309–330 cases per 100,000 <5 years [18] Canada (Ontario): 22 cases per 100,000 <5 years [19] Northern Europe: 5.4 to 11.4 cases per 100,000 <5 years [20] |
| Age of onset (Peak) | 1–5 years [22] | 2–5 years (1–2 years) |
| Male to female ratio | 1/1 | 1.5/1 |
| Family clustering | <1% | 1% |
| Spiking fever | Yes almost 100% | Yes almost 100% |
| Skin rash | Diffuse macular, urticarial (75–80%) | Diffuse Macular, urticarial, scarlatiniform (80%) [23] Oedema and redness of the palms and soles (70%) [23] |
| Mucous lesions | Odynophagia (adults 66%) | Conjunctiva redness (89%) [23] Diffuse redness of the oral cavity (72%) [23] Strawberry tongue (56%) [23] Dryness of the lips (85%) [23] |
| Adenitis | Diffuse adenitis (42%) | Cervical adenitis (55%) [23] |
| Gastro-intestinal symptoms | Abdominal pain | Diarrhoea (60%) [23] |
| Hepato-splenomegaly | Children 20% | Hepatomegaly (56%) [23] |
| Arthritis | Persistent (25%) | Transient, and unusual |
| Heart involvement | Pericarditis (18%) Myocarditis Coronary aneurysms | Pericarditis (18%) [23] Myocarditis (3%) [23] Coronary aneurysms 25% of untreated patients |
| Macrophage activation syndrome | Apparent 10% Subclinical 40% | |
| Disease course | Monophasic with variable duration (40%) Polycyclic (10%) Persistent (50%) | Monophasic > 97% Recurrent (3%) |
| Response to treatment (yes/No) | [24] | |
| Corticoids | Yes | Yes |
| IV Ig | No | Yes |
| Anti TNF | Few | Yes |
| Anti IL-1 | Yes | Probably yes, still exploratory |
| Anti IL-6 | Yes | Unknown |
| Biomarker | Still’s Disease | Kawasaki Disease |
|---|---|---|
| CRP | Unspecific elevation | Unspecific elevation Very high levels are related to cardiac damage and IVIG resistance |
| Complete blood cell count | Polynucleosis, anaemia, and thrombocytosis are indicative of active disease Cytopenia are related to MAS | Polynucleosis, anaemia, and thrombocytosis are indicative of active disease Cytopenia are related to MAS |
| ↑ d-dimer, ↓ fibrinogen | Indicative of MAS | Indicative of MAS |
| LDH, AST, ALT | Associated with disease activity | Associated with disease activity |
| Ferritin | Elevated, useful for diagnosis | Variably elevated |
| IL-6 | Elevated, may be related with arthritis feature | Very high levels are related to IVIG resistance and coronary aneurysms |
| IL-1β | Associated with systemic symptoms | Elevated and related to cardiac damage |
| IL-18 | Elevated in active systemic JIA and much higher in MAS | Elevated together with IL1β in active disease |
| TNFα | Normal or variably elevated | Elevated in acute phase |
| INFγ | Elevated in active systemic JIA and MAS | Very high levels are related to IVIG resistance and coronary aneurysms |
| S100 A12 | Elevated in active systemic JIA and MAS | Elevated and corelating with disease activity |
| sRAGE * | Decreased during active disease | Decreased during active disease |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dusser, P.; Koné-Paut, I. Still’s Disease in the Constellation of Hyperinflammatory Syndromes: A Link with Kawasaki Disease? J. Clin. Med. 2021, 10, 3244. https://doi.org/10.3390/jcm10153244
Dusser P, Koné-Paut I. Still’s Disease in the Constellation of Hyperinflammatory Syndromes: A Link with Kawasaki Disease? Journal of Clinical Medicine. 2021; 10(15):3244. https://doi.org/10.3390/jcm10153244
Chicago/Turabian StyleDusser, Perrine, and Isabelle Koné-Paut. 2021. "Still’s Disease in the Constellation of Hyperinflammatory Syndromes: A Link with Kawasaki Disease?" Journal of Clinical Medicine 10, no. 15: 3244. https://doi.org/10.3390/jcm10153244
APA StyleDusser, P., & Koné-Paut, I. (2021). Still’s Disease in the Constellation of Hyperinflammatory Syndromes: A Link with Kawasaki Disease? Journal of Clinical Medicine, 10(15), 3244. https://doi.org/10.3390/jcm10153244