
NF1-Dependent Transcriptome Regulation in the Melanocyte Lineage and in Melanoma



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. RNA Sequencing and Analysis
2.3. Data from cBioportal Database
3. Results and Discussion
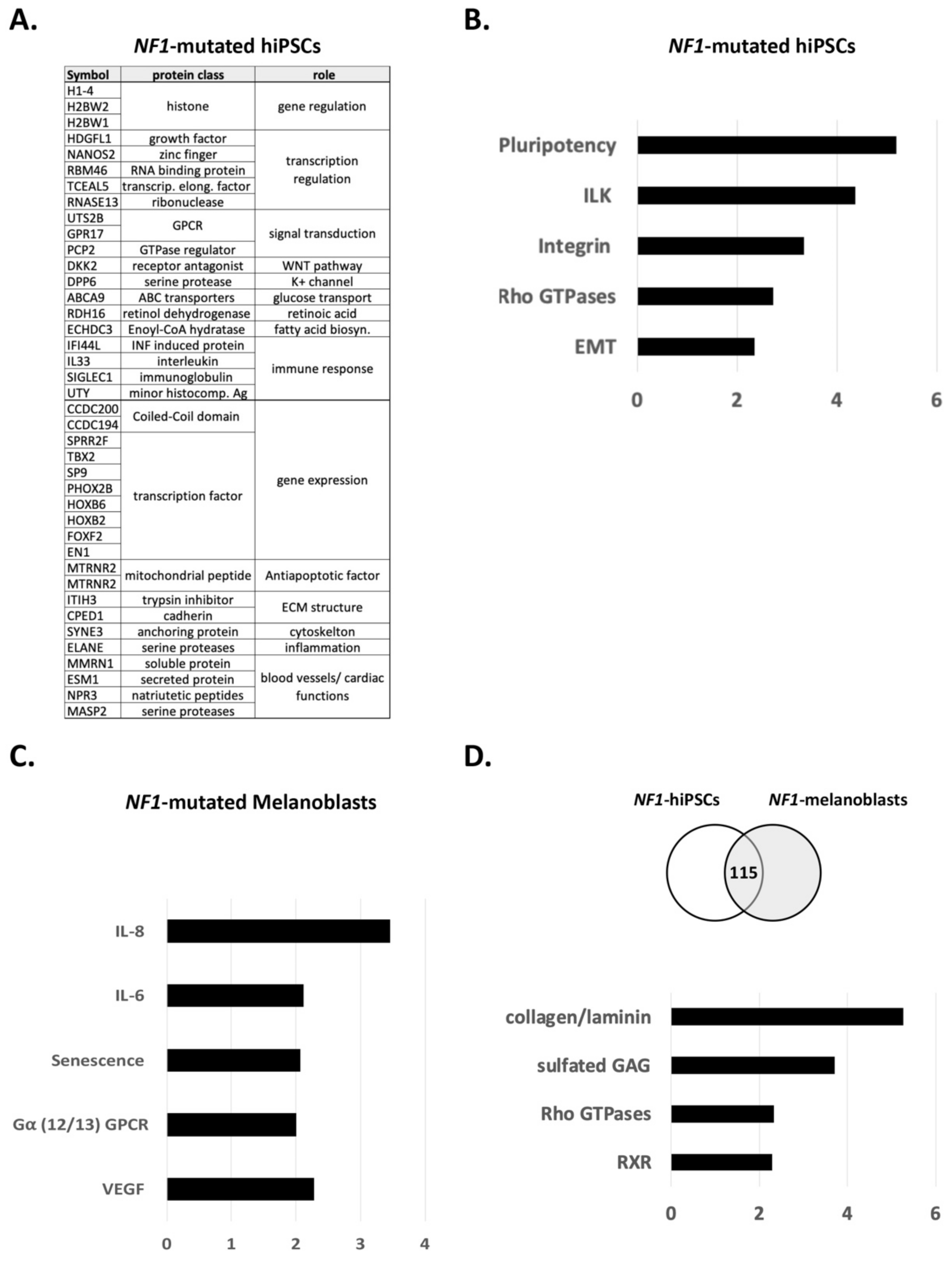
3.1. NF1 Mutations Have Differential Impact on Pluripotency and on Differentiation
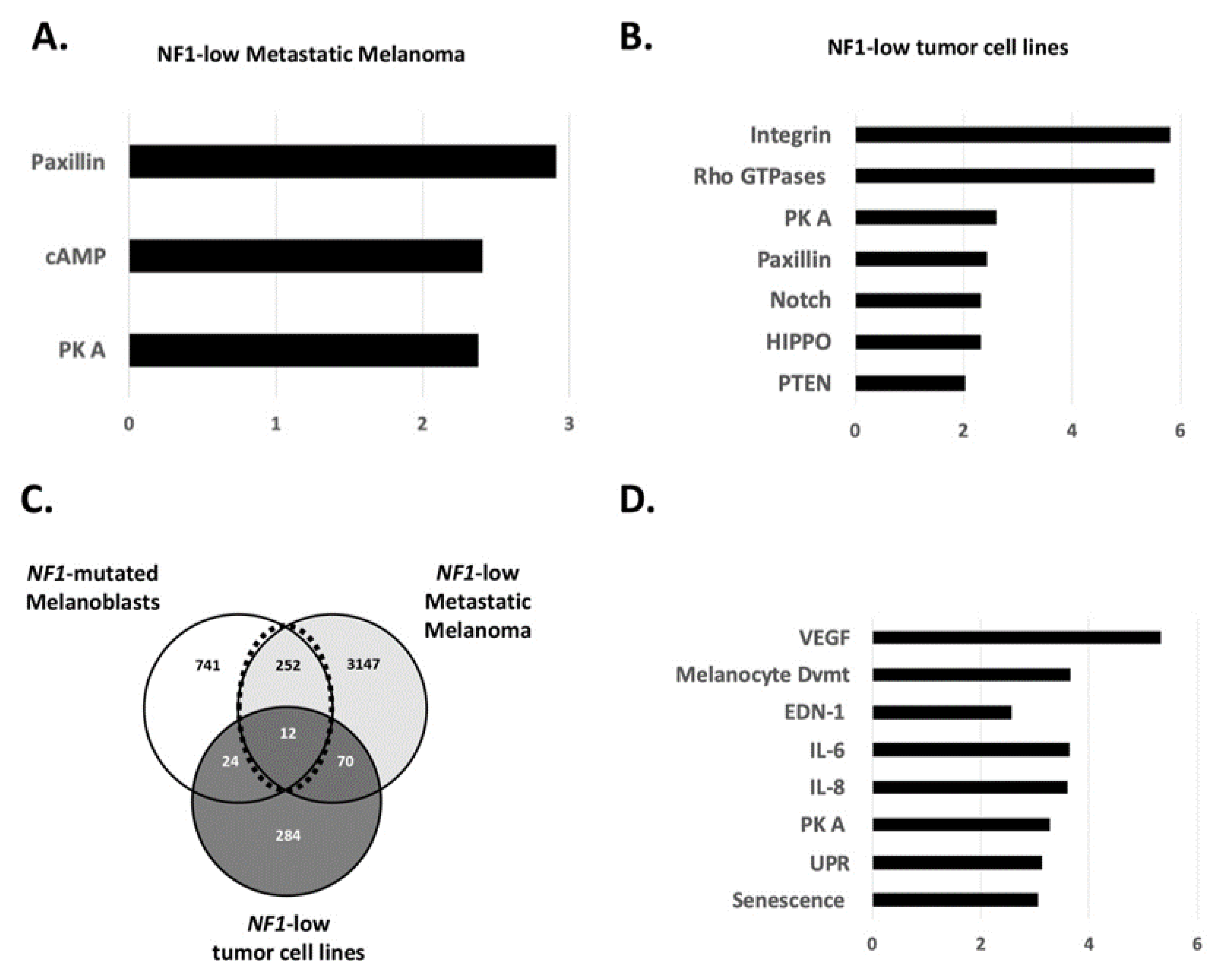
3.2. Role of NF1 in the Gene’s Regulation of Melanoblasts and Melanoma
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larribère, L.; Kuphal, S.; Sachpekidis, C.; Sachindra; Hüser, L.; Bosserhoff, A.; Utikal, J. Targeted Therapy-Resistant Melanoma Cells Acquire Transcriptomic Similarities with Human Melanoblasts. Cancers 2018, 10, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferner, R.E. Neurofibromatosis 1. Eur. J. Hum. Genet. 2007, 15, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Deo, M.; Huang, J.L.-Y.; Fuchs, H.; de Angelis, M.H.; Van Raamsdonk, C.D. Differential effects of neurofibromin gene dosage on melanocyte development. J. Investig. Dermatol. 2013, 133, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, M.; Huang, J.L.-Y.; Van Raamsdonk, C.D. Genetic interactions between neurofibromin and endothelin receptor B in mice. PLoS ONE 2013, 8, e59931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwakar, G.; Zhang, D.; Jiang, S.; Hornyak, T.J. Neurofibromin as a regulator of melanocyte development and differentiation. J. Cell Sci. 2008, 121, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrle-Haller, B.; Meller, M.; Weston, J.A. Analysis of melanocyte precursors in Nf1 mutants reveals that MGF/KIT signaling promotes directed cell migration independent of its function in cell survival. Dev. Biol. 2001, 232, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Birch, P.H. Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1728 patients. Am. J. Med. Genet. 1997, 70, 138–143. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Aylsworth, A.; Carey, J.C.; Korf, B.; Marks, J.; Pyeritz, R.E.; Rubenstein, A.; Viskochil, D. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997, 278, 51–57. [Google Scholar] [CrossRef]
- De Schepper, S.; Maertens, O.; Callens, T.; Naeyaert, J.-M.; Lambert, J.; Messiaen, L. Somatic mutation analysis in NF1 café au lait spots reveals two NF1 hits in the melanocytes. J. Investig. Dermatol. 2008, 128, 1050–1053. [Google Scholar] [CrossRef]
- Bos, J.L. Ras Oncogenes in Human Cancer: A Review ras Oncogenes in Human Cancer: A Review. Cancer Rese 1989, 49, 4682–4689. [Google Scholar]
- Powell, M.B.; Hyman, P.; Bell, O.D.; Balmain, A.; Brown, K.; Alberts, D.; Bowden, G.T. Hyperpigmentation and melanocytic hyperplasia in transgenic mice expressing the human T24 Ha-ras gene regulated by a mouse tyrosinase promoter. Mol. Carcinog. 1995, 12, 82–90. [Google Scholar] [CrossRef]
- Hemesath, T.J.; Price, E.R.; Takemoto, C.; Badalian, T.; Fisher, D.E. MAP kinase links the transcription factor Microphthalmia to c-Kit signalling in melanocytes. Nature 1998, 391, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, C.; Abbe, P.; Hemesath, T.J.; Bille, K.; Fisher, D.E.; Ortonne, J.P.; Ballotti, R. Microphthalmia gene product as a signal transducer in cAMP-induced differentiation of melanocytes. J. Cell Biol. 1998, 142, 827–835. [Google Scholar] [CrossRef] [Green Version]
- Larribère, L.; Cakrapradipta Wibowo, Y.; Patil, N.; Abba, M.; Tundidor, I.; Aguiñón Olivares, R.G.; Allgayer, H.; Utikal, J. NF1-RAC1 axis regulates migration of the melanocytic lineage. Transl. Oncol. 2020, 13, 100858. [Google Scholar] [CrossRef]
- Gregory, P.E.; Gutmann, D.H.; Mitchell, A.; Park, S.; Boguski, M.; Jacks, T.; Wood, D.L.; Jove, R.; Collins, F.S. Neurofibromatosis type 1 gene product (neurofibromin) associates with microtubules. Somat. Cell Mol. Genet. 1993, 19, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-L.; Lei, Y.-T.; Hong, C.-J.; Hsueh, Y.-P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841. [Google Scholar] [CrossRef] [Green Version]
- Kweha, F.; Zheng, M.; Kurenovaa, E.; Wallacec, M.; Golubovskayad, V.; Cance, W.G. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 2009, 48, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.J.; Erickson, C.A. The making of a melanocyte: The specification of melanoblasts from the neural crest. Pigment Cell Melanoma Res. 2008, 21, 598–610. [Google Scholar] [CrossRef]
- Lugassy, C.; Lazar, V.; Dessen, P.; van den Oord, J.J.; Winnepenninckx, V.; Spatz, A.; Bagot, M.; Bensussan, A.; Janin, A.; Eggermont, A.M.; et al. Gene expression profiling of human angiotropic primary melanoma: Selection of 15 differentially expressed genes potentially involved in extravascular migratory metastasis. Eur. J. Cancer 2011, 47, 1267–1275. [Google Scholar] [CrossRef]
- Lugassy, C.; Zadran, S.; Bentolila, L.A.; Wadehra, M.; Prakash, R.; Carmichael, S.T.; Kleinman, H.K.; Péault, B.; Larue, L.; Barnhill, R.L. Angiotropism, Pericytic Mimicry and Extravascular Migratory Metastasis in Melanoma: An Alternative to Intravascular Cancer Dissemination. Cancer Microenviron. 2014, 7, 139–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larribere, L.; Utikal, J. De- and re-differentiation of the melanocytic lineage. Eur. J. Cell Biol. 2013, 93, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Larribère, L.; Utikal, J. Stem Cell-Derived Models of Neural Crest Are Essential to Understand Melanoma Progression and Therapy Resistance. Front. Mol. Neurosci. 2019, 12, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linck-Paulus, L.; Lämmerhirt, L.; Völler, D.; Meyer, K.; Engelmann, J.C.; Spang, R.; Eichner, N.; Meister, G.; Kuphal, S.; Bosserhoff, A.K. Learning from Embryogenesis—A Comparative Expression Analysis in Melanoblast Differentiation and Tumorigenesis Reveals miRNAs Driving Melanoma Development. J. Clin. Med. 2021, 10, 2259. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.; Aksoy, A. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krauthammer, M.; Kong, Y.; Bacchiocchi, A.; Evans, P.; Pornputtapong, N.; Wu, C.; Mccusker, J.P.; Ma, S.; Cheng, E.; Straub, R.; et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat. Genet. 2015, 47, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Törngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1 -mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Colombino, M.; Casula, M.; Manca, A.; Mandalà, M.; Cossu, A. Molecular Pathways in Melanomagenesis: What We Learned from Next-Generation Sequencing Approaches. Curr. Oncol. Rep. 2018, 20, 86. [Google Scholar] [CrossRef] [Green Version]
- Nissan, M.H.; Pratilas, C.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Theurillat, J.-P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef] [Green Version]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for NF1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef] [Green Version]
- Larribere, L.; Wu, H.; Novak, D.; Galach, M.; Bernhardt, M.; Orouji, E.; Weina, K.; Knappe, N.; Sachpekidis, C.; Umansky, L.; et al. NF1 loss induces senescence during human melanocyte differentiation in an iPSC-based model. Pigment Cell Melanoma Res. 2015, 28, 407–416. [Google Scholar] [CrossRef]
- Brannan, C.I.; Perkins, A.S.; Vogel, K.S.; Ratner, N.; Nordlund, M.L.; Reid, S.W.; Buchberg, A.M.; Jenkins, N.A.; Parada, L.F.; Copeland, N.G. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994, 8, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossahebi-Mohammadi, M.; Quan, M.; Zhang, J.S.; Li, X. FGF Signaling Pathway: A Key Regulator of Stem Cell Pluripotency. Front. Cell Dev. Biol. 2020, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Spurlock, G.; Thomas, L.; Thomas, N.S.T.; Richards, M.; Mautner, V.-F.; Cooper, D.N.; Guha, A.; Yan, J. Microarray-based copy number analysis of neurofibromatosis type-1 (NF1)-associated malignant peripheral nerve sheath tumors reveals a role for Rho-GTPase pathway genes in NF1 tumorigenesis. Hum. Mutat. 2012, 33, 763–776. [Google Scholar] [CrossRef]
- Shu, X.; Pei, D. The function and regulation of mesenchymal-to-epithelial transition in somatic cell reprogramming. Curr. Opin. Genet. Dev. 2014, 28, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Hayashi, H.; Kamata, K.; Goto, T.M.; Sasaki, M.; Kuramochi, A.; Saya, H. Decreased expression of neurofibromin contributes to epithelial-mesenchymal transition in neurofibromatosis type 1. Exp. Dermatol. 2010, 19, e136–e141. [Google Scholar] [CrossRef]
- Kim, J.; Novak, D.; Sachpekidis, C.; Utikal, J.; Larribère, L. STAT3 Relays a Differential Response to Melanoma-Associated NRAS Mutations. Cancers 2020, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Allouche, J.; Bellon, N.; Saidani, M.; Stanchina-Chatrousse, L.; Masson, Y.; Patwardhan, A.; Gilles-Marsens, F.; Delevoye, C.; Domingues, S.; Nissan, X.; et al. In vitro modeling of hyperpigmentation associated to neurofibromatosis type 1 using melanocytes derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 201501032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Hudson, F.Z.; Xu, Z.; Tritz, R.; Rojas, M.; Patel, C.; Haigh, S.B.; Bordán, Z.; Ingram, D.A.; Fulton, D.J.; et al. Neurofibromin deficiency induces endothelial cell proliferation and retinal neovascularization. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2520–2528. [Google Scholar] [CrossRef]
- Worzfeld, T.; Wettschureck, N.; Offermanns, S. G12/G13-mediated signalling in mammalian physiology and disease. Trends Pharmacol. Sci. 2008, 29, 582–589. [Google Scholar] [CrossRef]
- Kedzierski, R.M.; Yanagisawa, M. Endothelin system: The double-edged sword in health and disease. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 851–876. [Google Scholar] [CrossRef] [PubMed]
- Bouallegue, A.; Bou Daou, G.; Srivastava, A. Endothelin-1-Induced Signaling Pathways in Vascular Smooth Muscle Cells. Curr. Vasc. Pharmacol. 2006, 5, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Sugden, P.; Clerk, A. Endothelin Signalling in the Cardiac Myocyte and its Pathophysiological Relevance. Curr. Vasc. Pharmacol. 2005, 3, 343–351. [Google Scholar] [CrossRef]
- Larribere, L.; Utikal, J. Update on GNA Alterations in Cancer: Implications for Uveal Melanoma Treatment. Cancers 2020, 12, 1524. [Google Scholar] [CrossRef]
- Larribere, L.; Utikal, J. Multiple roles of NF1 in the melanocyte lineage. Pigment Cell Melanoma Res. 2016. [Google Scholar] [CrossRef]
- Rossant, J. Mouse mutants and cardiac development: New molecular insights into cardiogenesis. Circ. Res. 1996, 78, 349–353. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, C.I.; Setaluri, V. Cyclic AMP (cAMP) signaling in melanocytes and melanoma. Arch. Biochem. Biophys. 2014, 563, 22–27. [Google Scholar] [CrossRef]
- Holcomb, N.C.; Bautista, R.M.; Jarrett, S.G.; Carter, K.M.; Gober, M.K.; D’Orazio, J.A. cAMP-mediated regulation of melanocyte genomic instability: A melanoma-preventive strategy. In Advances in Protein Chemistry and Structural Biology; Academic Press Inc.: Cambridge, MA, USA, 2019; Volume 115, pp. 247–295. [Google Scholar]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natale, C.A.; Li, J.; Zhang, J.; Dahal, A.; Dentchev, T.; Stanger, B.Z.; Ridky, T.W. Activation of G protein-coupled estrogen receptor signaling inhibits melanoma and improves response to immune checkpoint blockade. eLife 2018, 7, e31770. [Google Scholar] [CrossRef]
- Ranzani, M.; Alifrangis, C.; Perna, D.; Dutton-Regester, K.; Pritchard, A.; Wong, K.; Rashid, M.; Robles-Espinoza, C.D.; Hayward, N.K.; McDermott, U.; et al. BRAF/NRAS wild-type melanoma, NF1 status and sensitivity to trametinib. Pigment Cell Melanoma Res. 2015, 28, 117–119. [Google Scholar] [CrossRef] [Green Version]
- Hamamura, K.; Furukawa, K.; Hayashi, T.; Hattori, T.; Nakano, J.; Nakashima, H.; Okuda, T.; Mizutani, H.; Hattori, H.; Ueda, M.; et al. Ganglioside GD3 promotes cell growth and invasion through p130Cas and paxillin in malignant melanoma cells. Proc. Natl. Acad. Sci. USA 2005, 102, 11041–11046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Rao, P.K.; Wen, R.; Song, Y.; Muir, D.; Wallace, P.; Van Horne, S.J.; Tennekoon, G.I.; Kadesch, T. Notch and Schwann cell transformation. Oncogene 2004, 23, 1146–1152. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Mo, J.; Brosseau, J.P.; Shipman, T.; Wang, Y.; Liao, C.P.; Cooper, J.M.; Allaway, R.J.; Gosline, S.J.C.; Guinney, J.; et al. Spatiotemporal loss of NF1 in schwann cell lineage leads to different types of cutaneous neurofibroma susceptible to modification by the hippo pathway. Cancer Discov. 2019, 9, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keng, V.W.; Rahrmann, E.P.; Watson, A.L.; Tschida, B.R.; Moertel, C.L.; Jessen, W.J.; Rizvi, T.A.; Collins, M.H.; Ratner, N.; Largaespada, D.A. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 2012, 72, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fukunaga-Kalabis, M.; Li, L.; Herlyn, M. Developmental pathways activated in melanocytes and melanoma. Arch. Biochem. Biophys. 2014, 563, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, 415–425. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Walton, Z.; Yecies, J.L.; Li, D.; Chen, Y.; Malone, C.F.; Maertens, O.; Jeong, S.M.; Bronson, R.T.; Lebleu, V.; et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell 2011, 20, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, C.S.; Sun, B.C.; Dong, X.Y.; Sun, T.; Zhao, N.; Liu, Y.R.; Gu, Q. Promoting melanoma growth and metastasis by enhancing VEGF expression. Wspolczesna Onkol. 2012, 16, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, F.; Longakit, A.; Huang, J.L.Y.; Van Raamsdonk, C.D. Endothelin signaling promotes melanoma tumorigenesis driven by constitutively active GNAQ. Pigment Cell Melanoma Res. 2020, 33, 834–849. [Google Scholar] [CrossRef]
- Schäfer, A.; Haenig, B.; Erupathil, J.; Strickner, P.; Sabato, D.; Welford, R.W.D.; Klaeylé, L.; Simon, E.; Krepler, C.; Brafford, P.; et al. Inhibition of endothelin-B receptor signaling synergizes with MAPK pathway inhibitors in BRAF mutated melanoma. Oncogene 2021, 40, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Ennen, M.; Eline Keime, C.; Gambi, G.; Kieny, A.; Coassolo, S.; Thibault-Carpentier, C.; Margerin-Schaller, F.; Davidson, G.; Vagne, C.; Lipsker, D.; et al. MITF-High and MITF-Low Cells and a Novel Subpopulation Expressing Genes of Both Cell States Contribute to Intra-and Intertumoral Heterogeneity of Primary Melanoma. Clin. Cancer Res. 2017, 23, 7097–7107. [Google Scholar] [CrossRef] [Green Version]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larribère, L.; Utikal, J. NF1-Dependent Transcriptome Regulation in the Melanocyte Lineage and in Melanoma. J. Clin. Med. 2021, 10, 3350. https://doi.org/10.3390/jcm10153350
Larribère L, Utikal J. NF1-Dependent Transcriptome Regulation in the Melanocyte Lineage and in Melanoma. Journal of Clinical Medicine. 2021; 10(15):3350. https://doi.org/10.3390/jcm10153350
Chicago/Turabian StyleLarribère, Lionel, and Jochen Utikal. 2021. "NF1-Dependent Transcriptome Regulation in the Melanocyte Lineage and in Melanoma" Journal of Clinical Medicine 10, no. 15: 3350. https://doi.org/10.3390/jcm10153350
APA StyleLarribère, L., & Utikal, J. (2021). NF1-Dependent Transcriptome Regulation in the Melanocyte Lineage and in Melanoma. Journal of Clinical Medicine, 10(15), 3350. https://doi.org/10.3390/jcm10153350