
Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
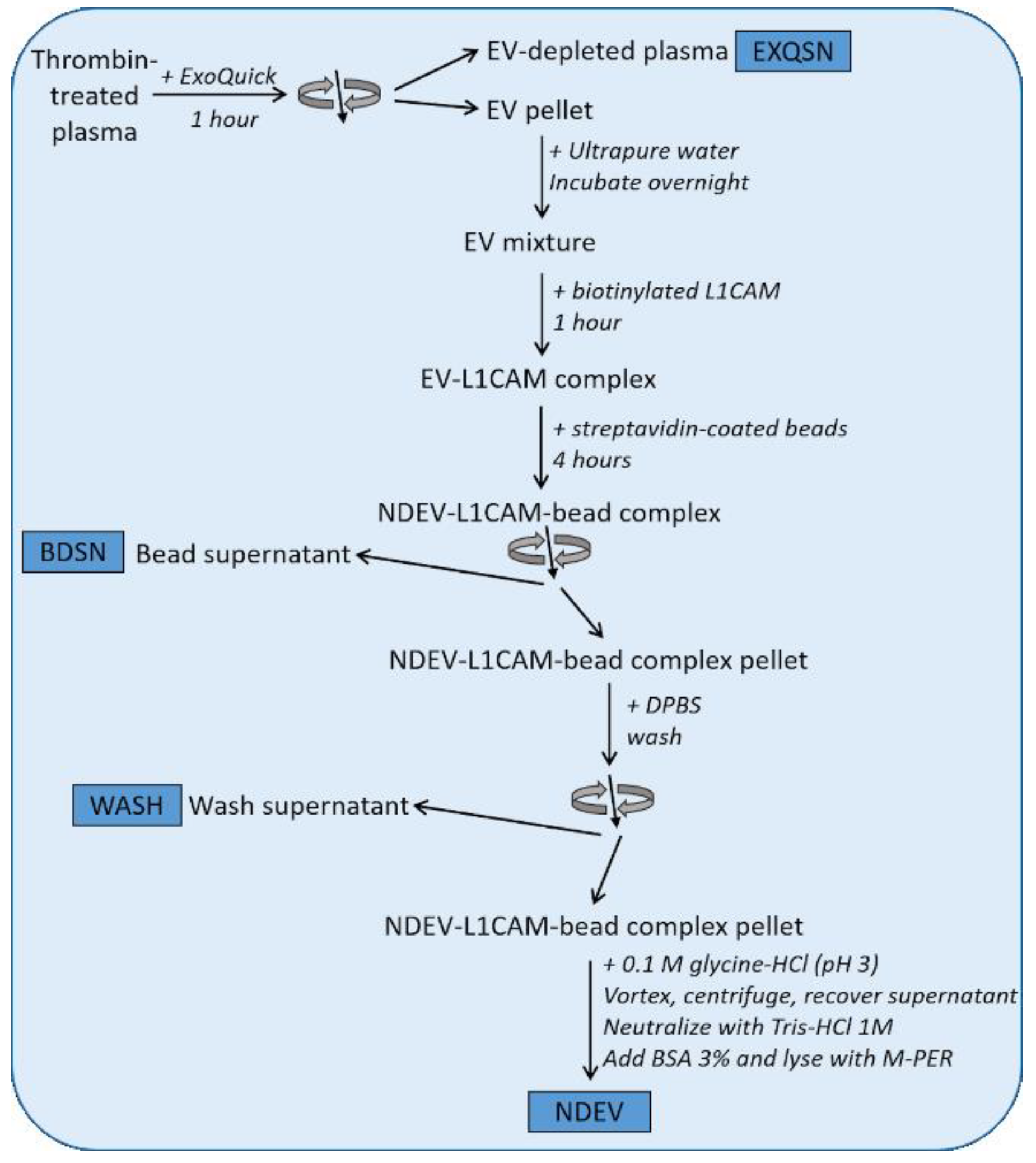
2.1. Isolation of NDEVs from Human Plasma
2.2. Validation of NDEVs
2.3. In Vitro Seeding and Conformational Tau Analysis
2.4. Stereotaxic Surgery
2.5. Immunohistochemistry and Image Analysis
2.6. Statistical Analyses
3. Results
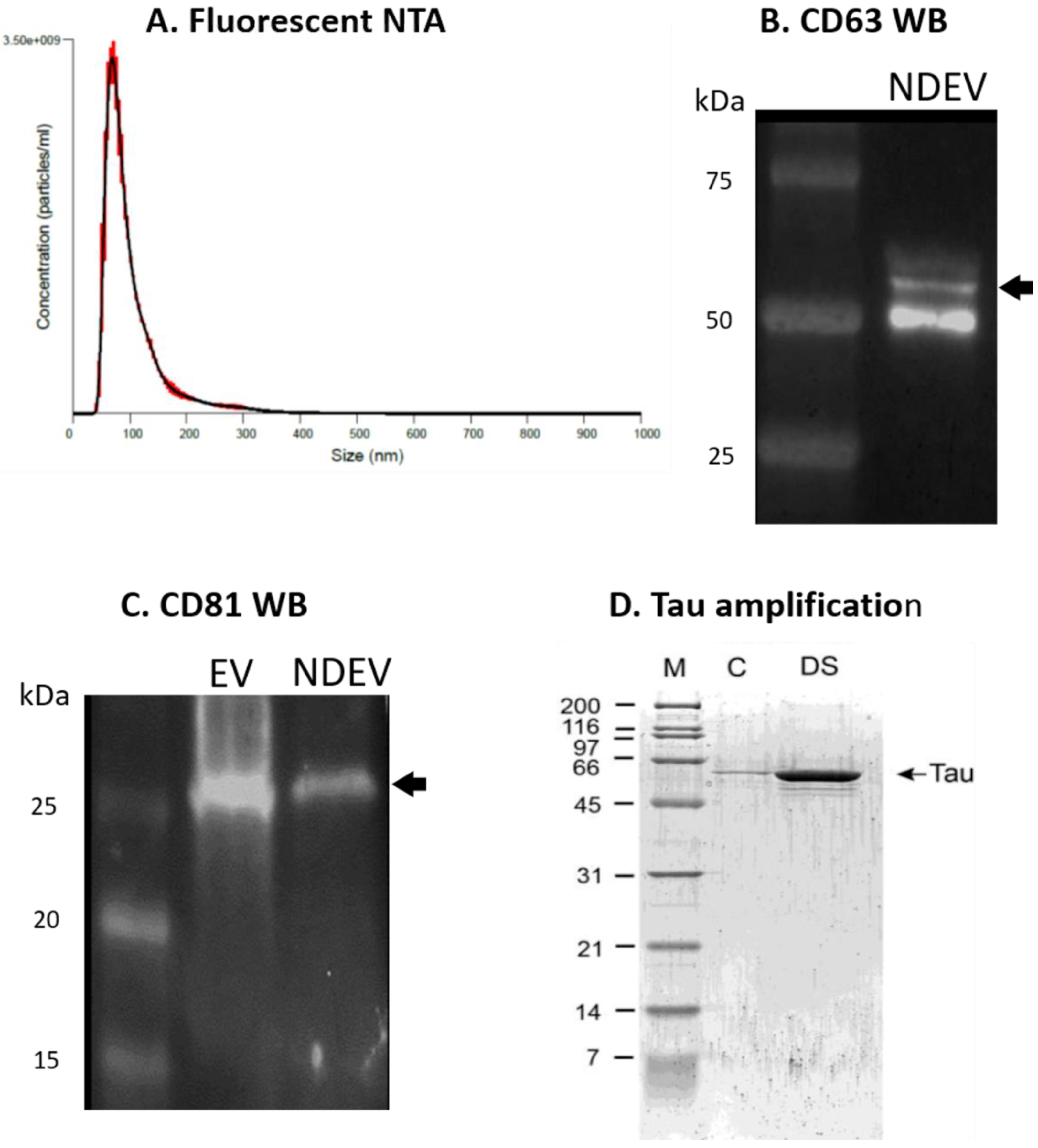
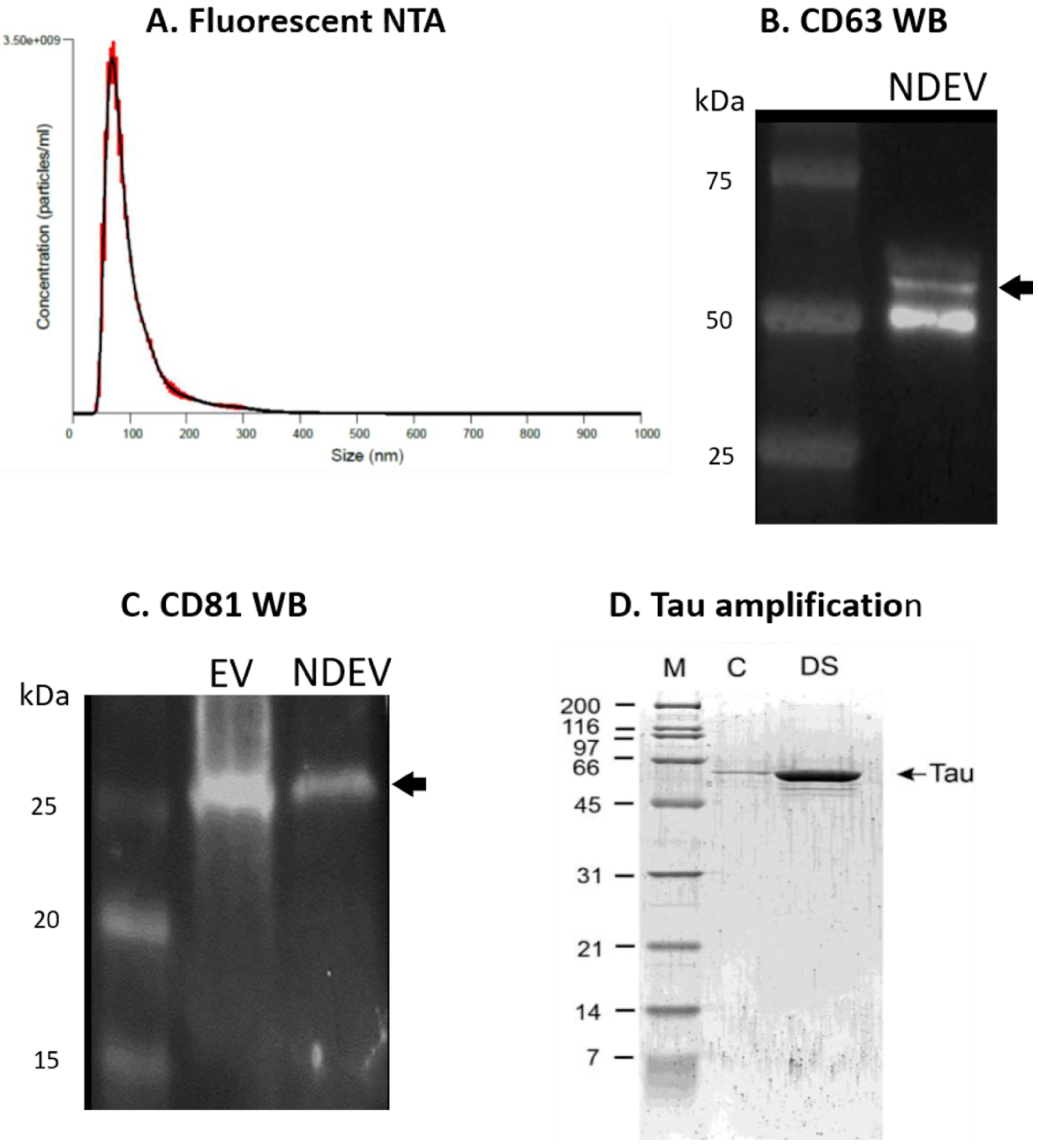
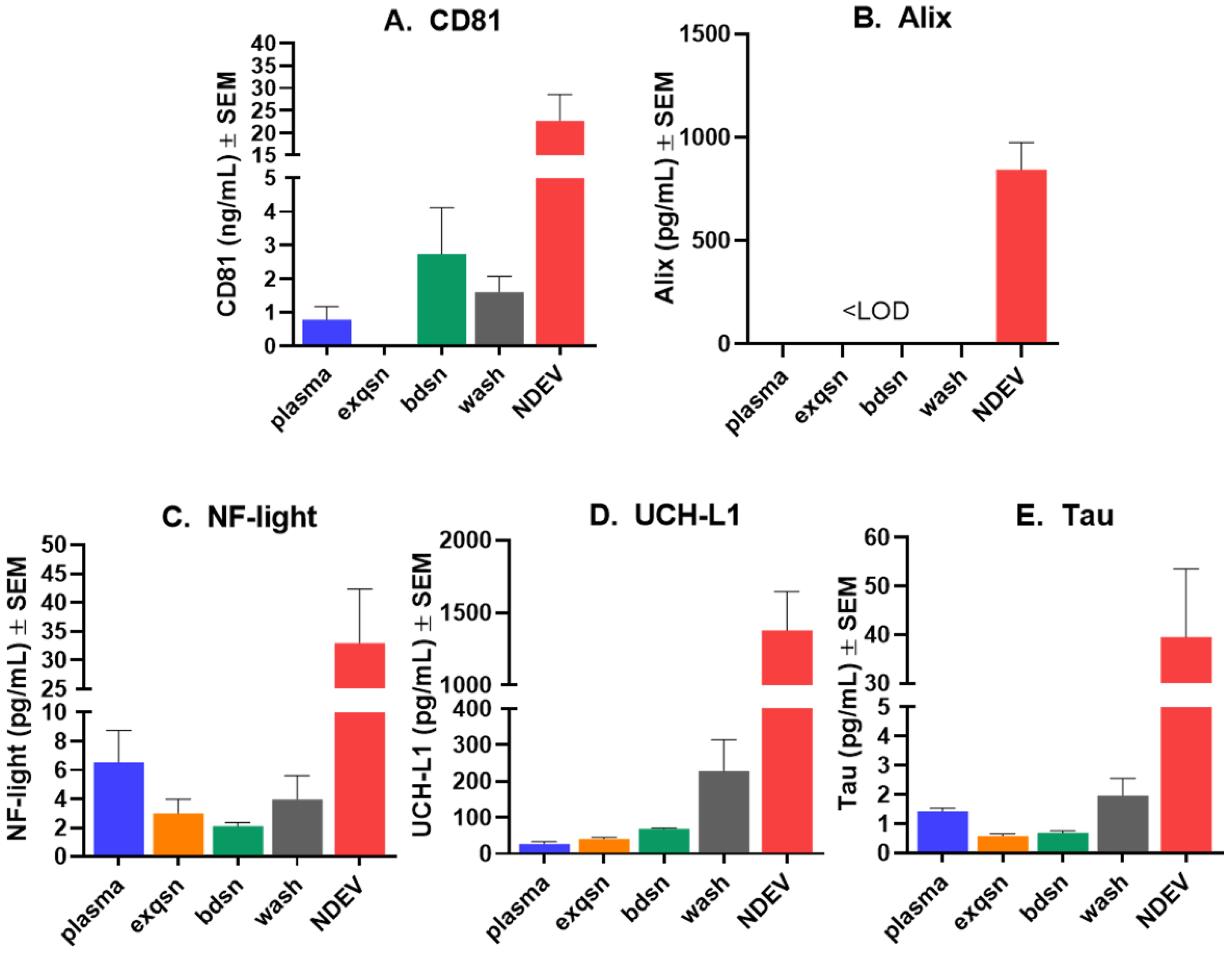
3.1. NDEV Validation and Characterization
3.2. Tau Protein Amplification and Aggregate Structure
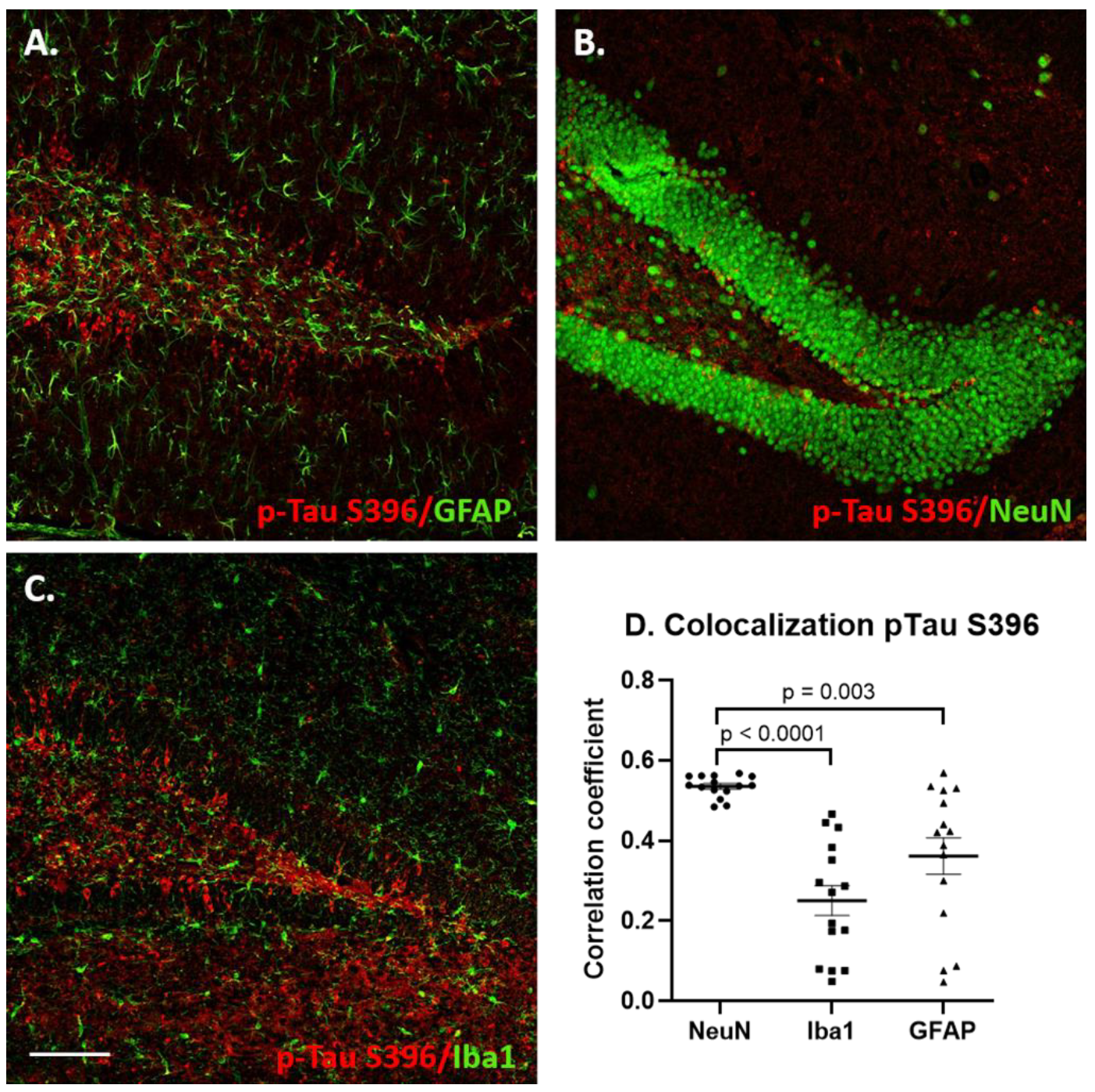
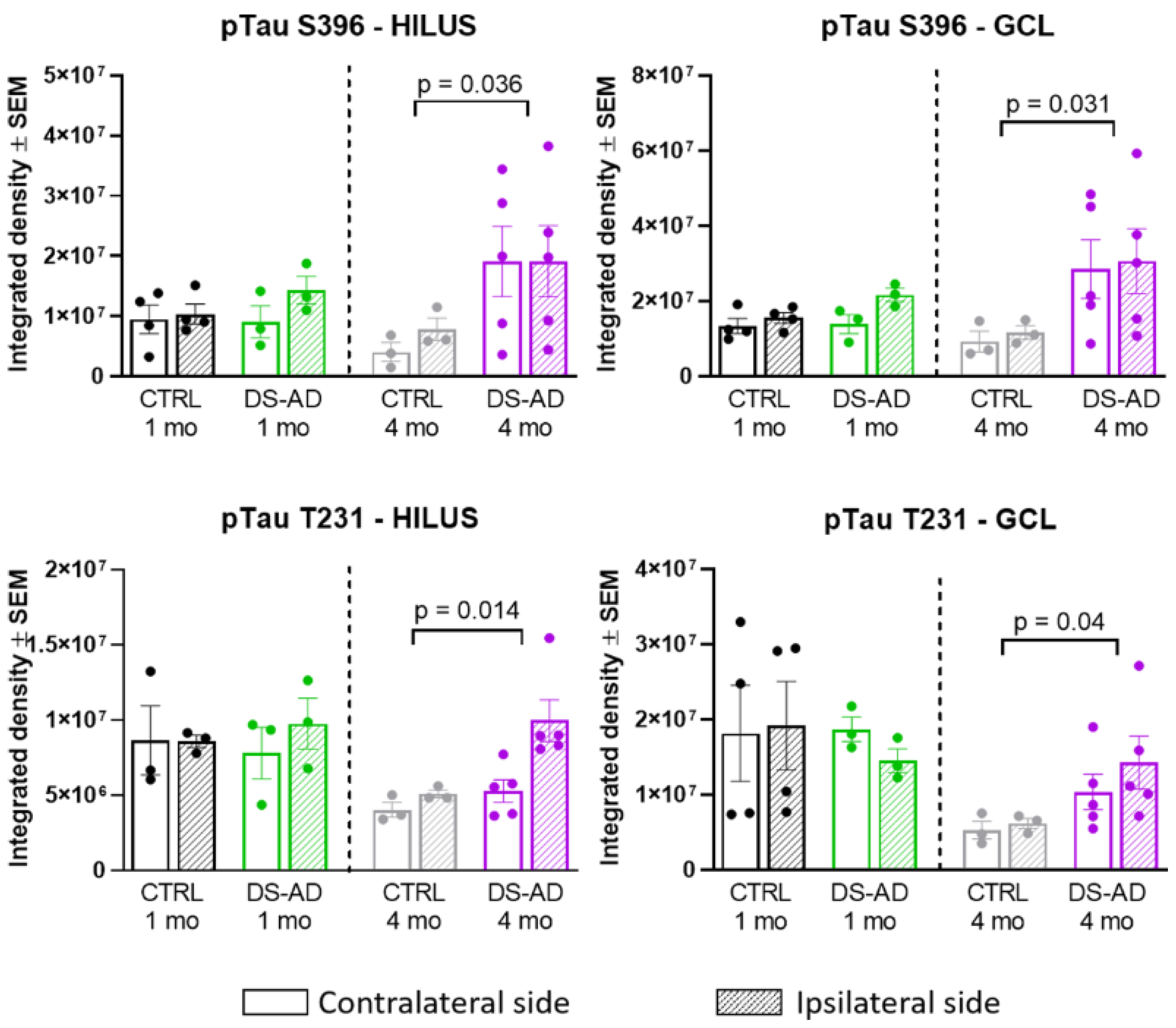
3.3. Increased p-Tau Staining Intensity 4 Months after the Injection
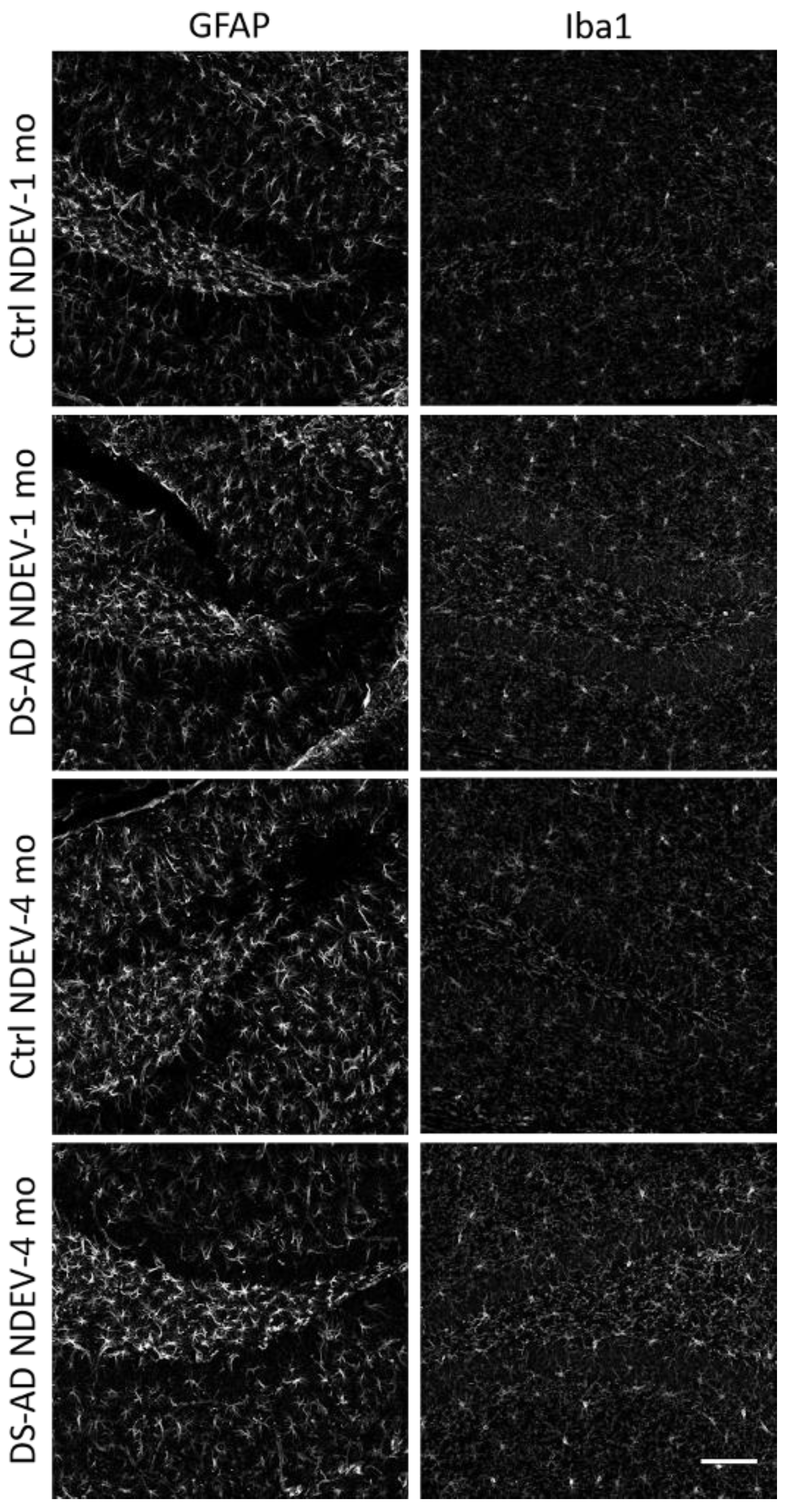
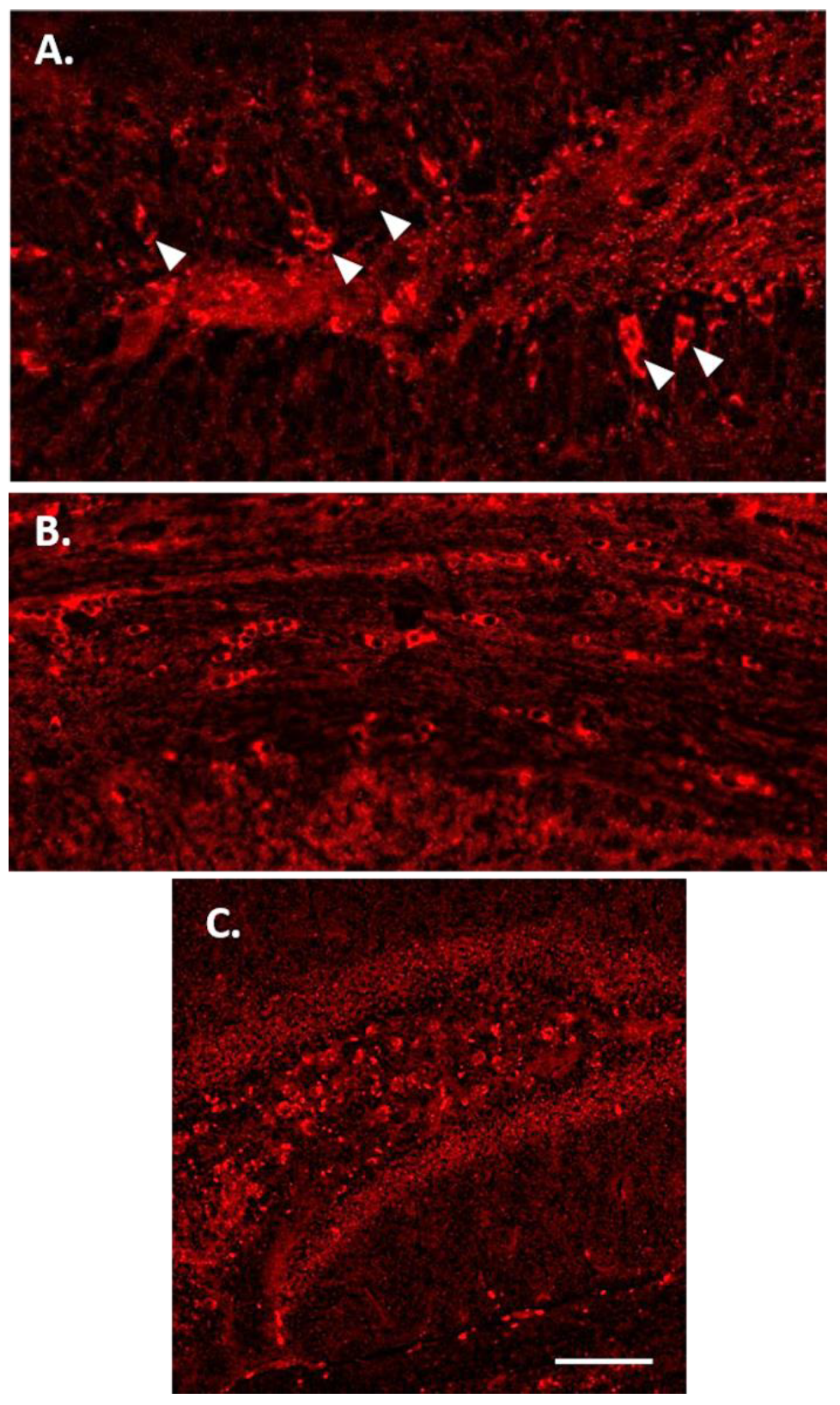
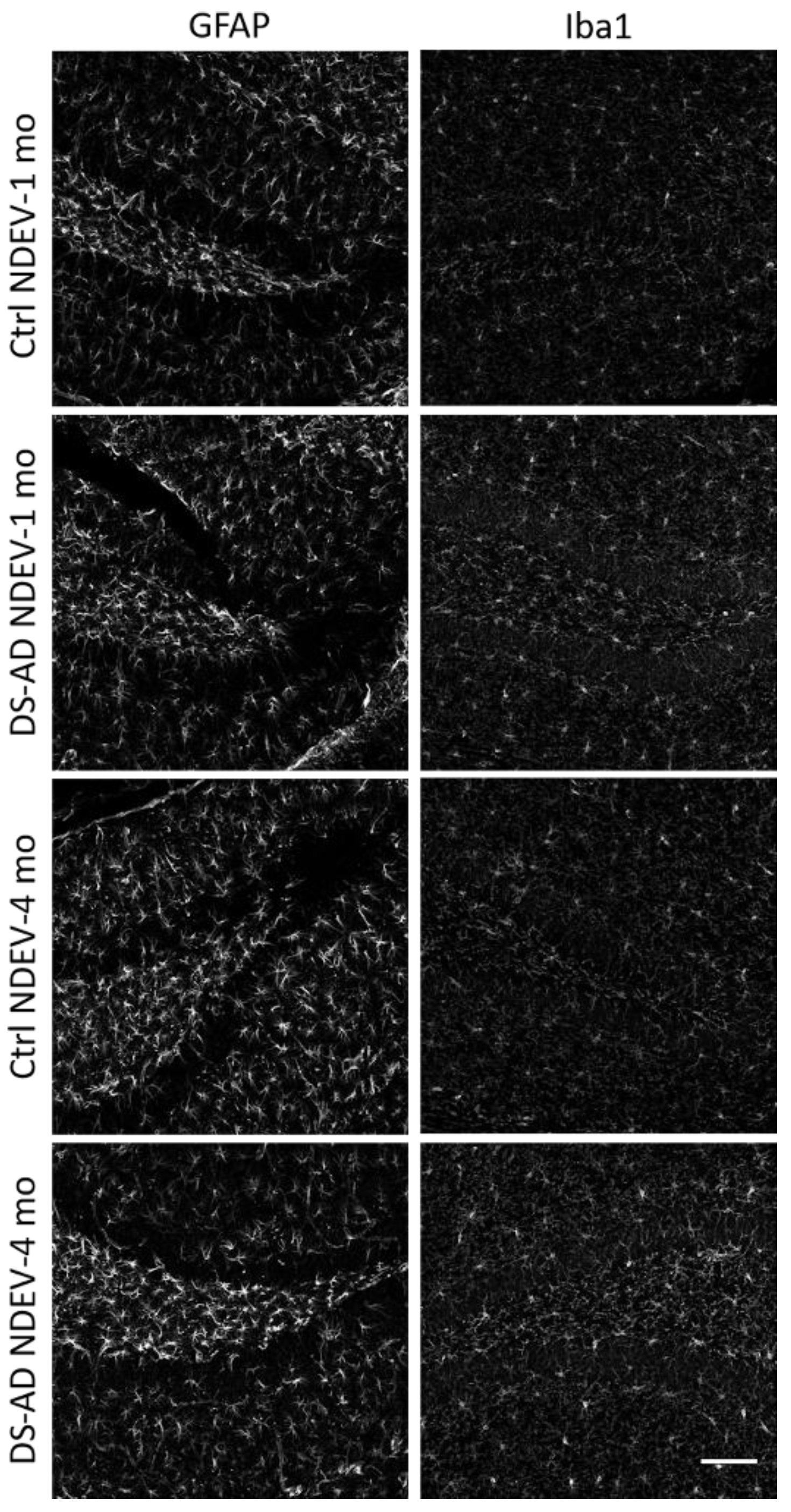
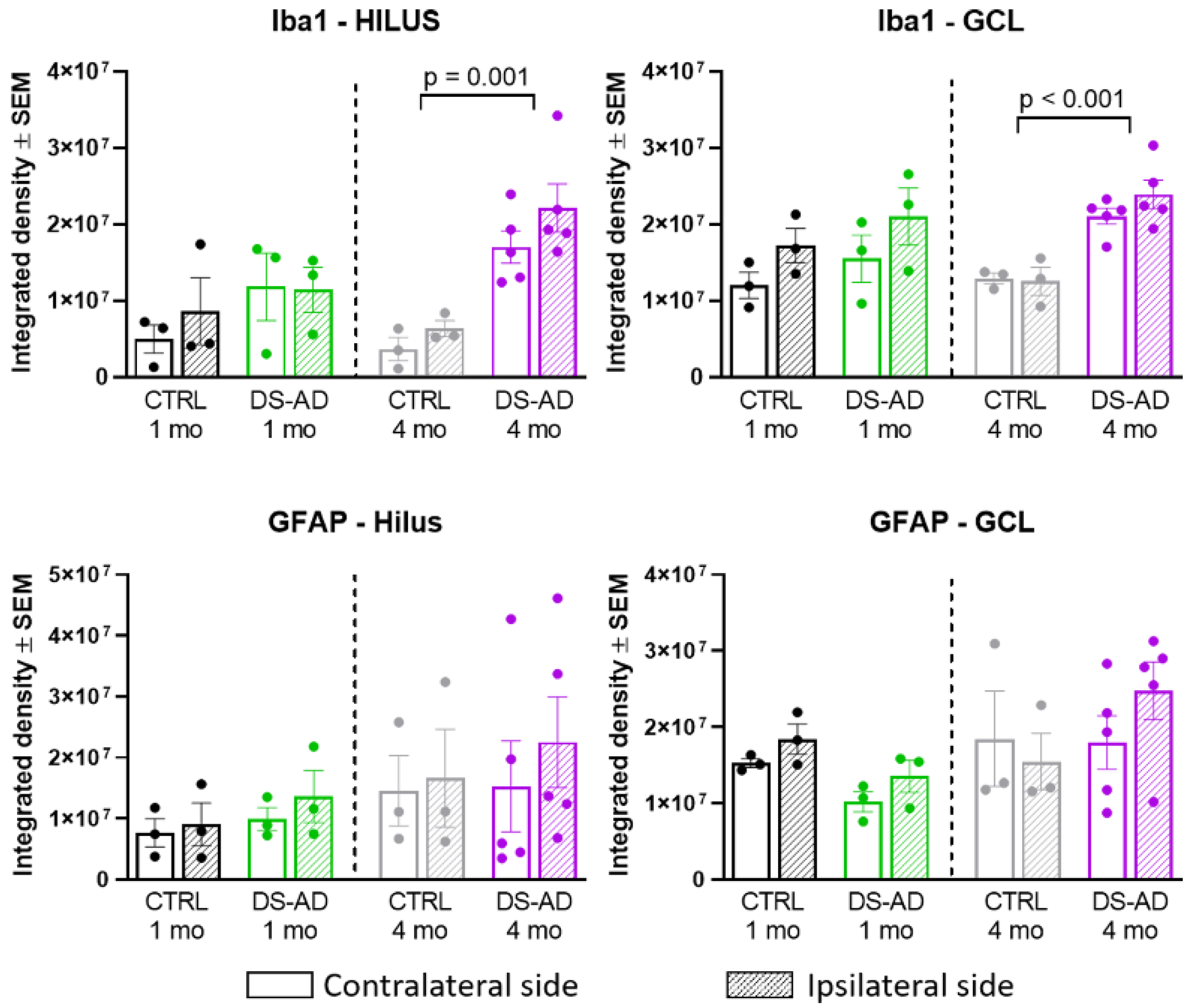
3.4. Glial Immunostaining
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Capone, G.T.; Chicoine, B.; Bulova, P.; Stephens, M.; Hart, S.; Crissman, B.; Videlefsky, A.; Myers, K.; Roizen, N.; Esbensen, A.; et al. Co-occurring medical conditions in adults with Down syndrome: A systematic review toward the development of health care guidelines. Am. J. Med. Genet. A 2018, 176, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Asim, A.; Kumar, A.; Muthuswamy, S.; Jain, S.; Agarwal, S. Down syndrome: An insight of the disease. J. Biomed. Sci. 2015, 22, 41. [Google Scholar] [CrossRef] [Green Version]
- Delabar, J.M.; Allinquant, B.; Bianchi, D.; Blumenthal, T.; Dekker, A.; Edgin, J.; O’Bryan, J.; Dierssen, M.; Potier, M.C.; Wiseman, F.; et al. Changing Paradigms in Down Syndrome: The First International Conference of the Trisomy 21 Research Society. Mol. Syndr. 2016, 7, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Egensperger, R.; Weggen, S.; Ida, N.; Multhaup, G.; Schnabel, R.; Beyreuther, K.; Bayer, T.A. Reverse relationship between beta-amyloid precursor protein and beta-amyloid peptide plaques in Down’s syndrome versus sporadic/familial Alzheimer’s disease. Acta Neuropathol. 1999, 97, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Fortea, J.; Carmona-Iragui, M.; Benejam, B.; Fernandez, S.; Videla, L.; Barroeta, I.; Alcolea, D.; Pegueroles, J.; Munoz, L.; Belbin, O.; et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet Neurol. 2018, 17, 860–869. [Google Scholar] [CrossRef]
- Godridge, H.; Reynolds, G.P.; Czudek, C.; Calcutt, N.A.; Benton, M. Alzheimer-like neurotransmitter deficits in adult Down’s syndrome brain tissue. J. Neurol. Neurosurg. Psychiatry 1987, 50, 775–778. [Google Scholar] [CrossRef] [Green Version]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Flores Aguilar, L.; Hanna, M.; Wisniewski, T.; Granholm, A.C.; Buhusi, M.; Busciglio, J.; et al. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Rafii, M.S.; Santoro, S.L. Prevalence and Severity of Alzheimer Disease in Individuals With Down Syndrome. JAMA Neurol. 2019, 76, 142–143. [Google Scholar] [CrossRef]
- Glasson, E.J.; Dye, D.E.; Bittles, A.H. The triple challenges associated with age-related comorbidities in Down syndrome. J. Intellect. Disabil. Res. 2014, 58, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Arbones, M.L.; Thomazeau, A.; Nakano-Kobayashi, A.; Hagiwara, M.; Delabar, J.M. DYRK1A and cognition: A lifelong relationship. Pharmacol. Ther. 2019, 194, 199–221. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Duchon, A.; Manousopoulou, A.; Loaec, N.; Villiers, B.; Pani, G.; Karatas, M.; Mechling, A.E.; Harsan, L.A.; Limanton, E.; et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis. Model. Mech. 2018, 11, dmm035634. [Google Scholar] [CrossRef] [Green Version]
- Peiris, H.; Keating, D.J. The neuronal and endocrine roles of RCAN1 in health and disease. Clin. Exp. Pharm. Physiol. 2018, 45, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome. Alzheimers Dement. 2017, 13, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Head, E.; Lott, I.T.; Wilcock, D.M.; Lemere, C.A. Aging in Down Syndrome and the Development of Alzheimer’s Disease Neuropathology. Curr. Alzheimer Res. 2016, 13, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Merezhko, M.; Uronen, R.L.; Huttunen, H.J. The Cell Biology of Tau Secretion. Front. Mol. Neurosci. 2020, 13, 569818. [Google Scholar] [CrossRef]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef]
- Castellani, R.J.; Perry, G. Tau Biology, Tauopathy, Traumatic Brain Injury, and Diagnostic Challenges. J. Alzheimers Dis. 2019, 67, 447–467. [Google Scholar] [CrossRef] [Green Version]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.-M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2001, 103, 26–35. [Google Scholar] [CrossRef]
- Gerson, J.E.; Mudher, A.; Kayed, R. Potential mechanisms and implications for the formation of tau oligomeric strains. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 482–496. [Google Scholar] [CrossRef]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, V.; Dinkel, P.D.; Rickman Hager, E.; Margittai, M. Amplification of Tau fibrils from minute quantities of seeds. Biochemistry 2014, 53, 5804–5809. [Google Scholar] [CrossRef] [Green Version]
- Siddiqua, A.; Luo, Y.; Meyer, V.; Swanson, M.A.; Yu, X.; Wei, G.; Zheng, J.; Eaton, G.R.; Ma, B.; Nussinov, R.; et al. Conformational basis for asymmetric seeding barrier in filaments of three- and four-repeat tau. J. Am. Chem. Soc. 2012, 134, 10271–10278. [Google Scholar] [CrossRef] [PubMed]
- Weismiller, H.A.; Murphy, R.; Wei, G.; Ma, B.; Nussinov, R.; Margittai, M. Structural disorder in four-repeat Tau fibrils reveals a new mechanism for barriers to cross-seeding of Tau isoforms. J. Biol. Chem. 2018, 293, 17336–17348. [Google Scholar] [CrossRef] [Green Version]
- Furman, J.L.; Vaquer-Alicea, J.; White, C.L., 3rd; Cairns, N.J.; Nelson, P.T.; Diamond, M.I. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017, 133, 91–100. [Google Scholar] [CrossRef]
- Raj, A.; Kuceyeski, A.; Weiner, M. A network diffusion model of disease progression in dementia. Neuron 2012, 73, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.; Merchant-Borna, K.; Jeromin, A.; Livingston, W.; Bazarian, J. Acute plasma tau relates to prolonged return to play after concussion. Neurology 2017, 88, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Elahi, F.M.; Mustapic, M.; Kapogiannis, D.; Pryhoda, M.; Gilmore, A.; Gorgens, K.A.; Davidson, B.; Granholm, A.C.; Ledreux, A. Altered levels of plasma neuron-derived exosomes and their cargo proteins characterize acute and chronic mild traumatic brain injury. FASEB J. 2019, 33, 5082–5088. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Liu, C.Y.; Merkel, S.F.; Ramirez, S.H.; Tierney, R.T.; Langford, D. Blood biomarkers for brain injury: What are we measuring? Neurosci. Biobehav. Rev. 2016, 68, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, K.; Qu, B.X.; Lai, C.; Devoto, C.; Motamedi, V.; Walker, W.C.; Levin, H.S.; Nolen, T.; Wilde, E.A.; Diaz-Arrastia, R.; et al. Higher exosomal phosphorylated tau and total tau among veterans with combat-related repetitive chronic mild traumatic brain injury. Brain Inj. 2018, 32, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Hardy, J.; Zetterberg, H. Neurological consequences of traumatic brain injuries in sports. Mol. Cell Neurosci. 2015, 66, 114–122. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Ferrer, I.; Andres-Benito, P.; Sala-Jarque, J.; Gil, V.; Del Rio, J.A. Capacity for Seeding and Spreading of Argyrophilic Grain Disease in a Wild-Type Murine Model; Comparisons With Primary Age-Related Tauopathy. Front. Mol. Neurosci. 2020, 13, 101. [Google Scholar] [CrossRef]
- Ferrer, I.; Zelaya, M.V.; Aguilo Garcia, M.; Carmona, M.; Lopez-Gonzalez, I.; Andres-Benito, P.; Lidon, L.; Gavin, R.; Garcia-Esparcia, P.; Del Rio, J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol. 2020, 30, 298–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichou, Y.; Lin, Y.; Rauch, J.N.; Vigers, M.; Zeng, Z.; Srivastava, M.; Keller, T.J.; Freed, J.H.; Kosik, K.S.; Han, S. Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc. Natl. Acad. Sci. USA 2018, 115, 13234–13239. [Google Scholar] [CrossRef] [Green Version]
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau Oligomers as Pathogenic Seeds: Preparation and Propagation In Vitro and In Vivo. Methods Mol. Biol. 2017, 1523, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.; Mairet-Coello, G.; Danis, C.; Lieger, S.; Caillierez, R.; Carrier, S.; Skrobala, E.; Landrieu, I.; Michel, A.; Schmitt, M.; et al. Prevention of tau seeding and propagation by immunotherapy with a central tau epitope antibody. Brain 2019, 142, 1736–1750. [Google Scholar] [CrossRef]
- Arbo, B.D.; Cechinel, L.R.; Palazzo, R.P.; Siqueira, I.R. Endosomal dysfunction impacts extracellular vesicle release: Central role in Abeta pathology. Ageing Res. Rev. 2020, 58, 101006. [Google Scholar] [CrossRef] [PubMed]
- Badhwar, A.; Haqqani, A.S. Biomarker potential of brain-secreted extracellular vesicles in blood in Alzheimer’s disease. Alzheimers Dement. 2020, 12, e12001. [Google Scholar] [CrossRef]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.J.; Zhao, B.; Zhao, J.; Li, S. Potential Roles of Exosomal MicroRNAs as Diagnostic Biomarkers and Therapeutic Application in Alzheimer’s Disease. Neural Plast. 2017, 2017, 7027380. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2, 20389. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Didiot, M.C.; Sapp, E.; Leszyk, J.; Shaffer, S.A.; Rockwell, H.E.; Gao, F.; Narain, N.R.; DiFiglia, M.; Kiebish, M.A.; et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles 2016, 5, 32570. [Google Scholar] [CrossRef]
- Smolarz, M.; Pietrowska, M.; Matysiak, N.; Mielanczyk, L.; Widlak, P. Proteome Profiling of Exosomes Purified from a Small Amount of Human Serum: The Problem of Co-Purified Serum Components. Proteomes 2019, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Banizs, A.B.; Huang, T.; Nakamoto, R.K.; Shi, W.; He, J. Endocytosis Pathways of Endothelial Cell Derived Exosomes. Mol. Pharm. 2018, 15, 5585–5590. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKelvey, K.J.; Powell, K.L.; Ashton, A.W.; Morris, J.M.; McCracken, S.A. Exosomes: Mechanisms of Uptake. J. Circ. Biomark. 2015, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic analysis of microglia-derived exosomes: Metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J. Immunol. 2005, 175, 2237–2243. [Google Scholar] [CrossRef] [Green Version]
- Fruhbeis, C.; Frohlich, D.; Kuo, W.P.; Kramer-Albers, E.M. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell Neurosci. 2013, 7, 182. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Willis, C.M.; Menoret, A.; Jellison, E.R.; Nicaise, A.M.; Vella, A.T.; Crocker, S.J. A Refined Bead-Free Method to Identify Astrocytic Exosomes in Primary Glial Cultures and Blood Plasma. Front. Neurosci. 2017, 11, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winston, C.N.; Romero, H.K.; Ellisman, M.; Nauss, S.; Julovich, D.A.; Conger, T.; Hall, J.R.; Campana, W.; O’Bryant, S.E.; Nievergelt, C.M.; et al. Assessing Neuronal and Astrocyte Derived Exosomes From Individuals With Mild Traumatic Brain Injury for Markers of Neurodegeneration and Cytotoxic Activity. Front. Neurosci. 2019, 13, 1005. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Gomez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Kramer-Albers, E.M.; Bretz, N.; Tenzer, S.; Winterstein, C.; Mobius, W.; Berger, H.; Nave, K.A.; Schild, H.; Trotter, J. Oligodendrocytes secrete exosomes containing major myelin and stress-protective proteins: Trophic support for axons? Proteom. Clin. Appl. 2007, 1, 1446–1461. [Google Scholar] [CrossRef]
- Caruso Bavisotto, C.; Scalia, F.; Marino Gammazza, A.; Carlisi, D.; Bucchieri, F.; Conway de Macario, E.; Macario, A.J.L.; Cappello, F.; Campanella, C. Extracellular Vesicle-Mediated Cell(-)Cell Communication in the Nervous System: Focus on Neurological Diseases. Int. J. Mol. Sci. 2019, 20, 434. [Google Scholar] [CrossRef] [Green Version]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 2012, 3, 145. [Google Scholar] [CrossRef] [Green Version]
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.H.; Mably, A.J.; O’Dowd, S.T.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M.; et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Abeta assemblies in vivo. Mol. Brain 2013, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalani, A.; Tyagi, N. Exosomes in neurological disease, neuroprotection, repair and therapeutics: Problems and perspectives. Neural Regen. Res. 2015, 10, 1565–1567. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [Green Version]
- Quek, C.; Hill, A.F. The role of extracellular vesicles in neurodegenerative diseases. Biochem. Biophys. Res. Commun. 2017, 483, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Howitt, J.; Hill, A.F. Exosomes in the Pathology of Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 26589–26597. [Google Scholar] [CrossRef] [Green Version]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Gotz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjo, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. 2016, 3, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Z.; Pathak, D.; Venkatesan Kalavai, S.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2021, 144, 288–309. [Google Scholar] [CrossRef]
- Reilly, P.; Winston, C.N.; Baron, K.R.; Trejo, M.; Rockenstein, E.M.; Akers, J.C.; Kfoury, N.; Diamond, M.; Masliah, E.; Rissman, R.A.; et al. Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol. Dis. 2017, 106, 222–234. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-beta pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, V.A.; Devoto, C.; Leete, J.; Sass, D.; Acott, J.D.; Mithani, S.; Gill, J.M. Extracellular Vesicle Proteins and MicroRNAs as Biomarkers for Traumatic Brain Injury. Front. Neurol. 2020, 11, 663. [Google Scholar] [CrossRef]
- Huang, M.; Hong, Z.; Xiao, C.; Li, L.; Chen, L.; Cheng, S.; Lei, T.; Zheng, H. Effects of Exosomes on Neurological Function Recovery for Ischemic Stroke in Pre-clinical Studies: A Meta-analysis. Front. Cell Neurosci. 2020, 14, 593130. [Google Scholar] [CrossRef]
- Sterzenbach, U.; Putz, U.; Low, L.H.; Silke, J.; Tan, S.S.; Howitt, J. Engineered Exosomes as Vehicles for Biologically Active Proteins. Mol. Ther. 2017, 25, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Milenkovic, I.; Jarc, J.; Dassler, E.; Aronica, E.; Iyer, A.; Adle-Biassette, H.; Scharrer, A.; Reischer, T.; Hainfellner, J.A.; Kovacs, G.G. The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down syndrome. Neuropathol. Appl. Neurobiol. 2018, 44, 314–327. [Google Scholar] [CrossRef]
- Lemoine, L.; Ledreux, A.; Mufson, E.J.; Perez, S.E.; Simic, G.; Doran, E.; Lott, I.; Carroll, S.; Bharani, K.; Thomas, S.; et al. Regional binding of tau and amyloid PET tracers in Down syndrome autopsy brain tissue. Mol. Neurodegener. 2020, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Bi, J.; Huang, J.; Tang, Y.; Du, S.; Li, P. Exosome: A Review of Its Classification, Isolation Techniques, Storage, Diagnostic and Targeted Therapy Applications. Int. J. Nanomed. 2020, 15, 6917–6934. [Google Scholar] [CrossRef]
- Vogt, B.A.; Paxinos, G. Cytoarchitecture of mouse and rat cingulate cortex with human homologies. Brain Struct. Funct. 2014, 219, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Luna-Munoz, J.; Chavez-Macias, L.; Garcia-Sierra, F.; Mena, R. Earliest stages of tau conformational changes are related to the appearance of a sequence of specific phospho-dependent tau epitopes in Alzheimer’s disease. J. Alzheimers Dis. 2007, 12, 365–375. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schimid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharani, K.L.; Ledreux, A.; Gilmore, A.; Carroll, S.L.; Granholm, A.C. Serum pro-BDNF levels correlate with phospho-tau staining in Alzheimer’s disease. Neurobiol. Aging 2020, 87, 49–59. [Google Scholar] [CrossRef]
- Sun, B.; Dalvi, P.; Abadjian, L.; Tang, N.; Pulliam, L. Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS 2017, 31, F9. [Google Scholar] [CrossRef]
- Mustapic, M.; Eitan, E.; Werner, J.K., Jr.; Berkowitz, S.T.; Lazaropoulos, M.P.; Tran, J.; Goetzl, E.J.; Kapogiannis, D. Plasma Extracellular Vesicles Enriched for Neuronal Origin: A Potential Window into Brain Pathologic Processes. Front. Neurosci. 2017, 11, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkel, P.D.; Siddiqua, A.; Huynh, H.; Shah, M.; Margittai, M. Variations in filament conformation dictate seeding barrier beween three- and four-repeat tau. Biochemistry 2011, 50, 4330–4336. [Google Scholar] [CrossRef]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Begard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, S.A.; Perez-Gonzalez, R.; Sharma, A.; Huang, F.K.; Alldred, M.J.; Pawlik, M.; Kaur, G.; Ginsberg, S.D.; Neubert, T.A.; Levy, E. Enhanced exosome secretion in Down syndrome brain—A protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol. Commun. 2017, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Hamlett, E.D.; LaRosa, A.; Mufson, E.J.; Fortea, J.; Ledreux, A.; Granholm, A.C. Exosome release and cargo in Down syndrome. Dev. Neurobiol. 2019, 79, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Darji, S.; Sato, Y.; Mellen, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Colacurcio, D.J.; Pensalfini, A.; Jiang, Y.; Nixon, R.A. Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 2018, 114, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-betaCTF (C99). J. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef]
- de Jong, O.G.; van Balkom, B.W.; Gremmels, H.; Verhaar, M.C. Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2. J. Cell Mol. Med. 2016, 20, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Batool, A.; Hill, T.D.M.; Nguyen, N.T.; Langa, E.; Diviney, M.; Mooney, C.; Brennan, G.P.; Connolly, N.M.C.; Sanz-Rodriguez, A.; Cavanagh, B.L.; et al. Altered Biogenesis and MicroRNA Content of Hippocampal Exosomes Following Experimental Status Epilepticus. Front. Neurosci. 2019, 13, 1404. [Google Scholar] [CrossRef] [Green Version]
- Abdulrahman, B.A.; Abdelaziz, D.H.; Schatzl, H.M. Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem. 2018, 293, 8956–8968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Talavera, Y.; Benito, I.; Casanas, J.J.; Rodriguez-Moreno, A.; Montesinos, M.L. Rapamycin restores BDNF-LTP and the persistence of long-term memory in a model of Down’s syndrome. Neurobiol. Dis. 2015, 82, 516–525. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Hargash, T.; Pawlik, M.; Goulbourne, C.N.; Perez-Gonzalez, R.; Levy, E. Enhanced generation of intraluminal vesicles in neuronal late endosomes in the brain of a Down syndrome mouse model with endosomal dysfunction. Dev. Neurobiol. 2019, 79, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Perea, J.R.; Llorens-Martin, M.; Avila, J.; Bolos, M. The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front. Cell Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef] [Green Version]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2018, 19, 663. [Google Scholar] [CrossRef] [Green Version]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, A.; Ribeiro, A.R.; Monteiro, M.; Garcia, G.; Vaz, A.R.; Brites, D. Secretome from SH-SY5Y APPSwe cells trigger time-dependent CHME3 microglia activation phenotypes, ultimately leading to miR-21 exosome shuttling. Biochimie 2018, 155, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Pulliam, L. Exosomes as mediators of neuroinflammation. J. Neuroinflamm. 2014, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Spanic, E.; Langer Horvat, L.; Hof, P.R.; Simic, G. Role of Microglial Cells in Alzheimer’s Disease Tau Propagation. Front. Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef] [Green Version]
- Chalermpalanupap, T.; Weinshenker, D.; Rorabaugh, J.M. Down but Not Out: The Consequences of Pretangle Tau in the Locus Coeruleus. Neural Plast. 2017, 2017, 7829507. [Google Scholar] [CrossRef] [Green Version]
- Gasic-Milenkovic, J.; Dukic-Stefanovic, S.; Deuther-Conrad, W.; Gartner, U.; Munch, G. Beta-amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon -gamma and ‘advanced glycation endproducts’ in a murine microglia cell line. Eur. J. Neurosci. 2003, 17, 813–821. [Google Scholar] [CrossRef]
- Pascual, M.; Ibanez, F.; Guerri, C. Exosomes as mediators of neuron-glia communication in neuroinflammation. Neural Regen. Res. 2020, 15, 796–801. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Borgmann, K.; Edara, V.V.; Stacy, S.; Ghorpade, A.; Ikezu, T. Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J. Extracell. Vesicles 2020, 9, 1706801. [Google Scholar] [CrossRef] [PubMed]
- Farroni, C.; Marasco, E.; Marcellini, V.; Giorda, E.; Valentini, D.; Petrini, S.; D’Oria, V.; Pezzullo, M.; Cascioli, S.; Scarsella, M.; et al. Dysregulated miR-155 and miR-125b Are Related to Impaired B-cell Responses in Down Syndrome. Front. Immunol. 2018, 9, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huggard, D.; Doherty, D.G.; Molloy, E.J. Immune Dysregulation in Children With Down Syndrome. Front. Pediatr. 2020, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Manti, S.; Cutrupi, M.C.; Cuppari, C.; Ferro, E.; Dipasquale, V.; Di Rosa, G.; Chimenz, R.; La Rosa, M.A.; Valenti, A.; Salpietro, V. Inflammatory biomarkers and intellectual disability in patients with Down syndrome. J. Intellect. Disabil. Res. 2018, 62, 382–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nateghi Rostami, M.; Douraghi, M.; Miramin Mohammadi, A.; Nikmanesh, B. Altered serum pro-inflammatory cytokines in children with Down’s syndrome. Eur. Cytokine Netw. 2012, 23, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, D.M.; Griffin, W.S. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflamm. 2013, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jaber, V.; Percy, M.E.; Lukiw, W.J. A microRNA cluster (let-7c, miRNA-99a, miRNA-125b, miRNA-155 and miRNA-802) encoded at chr21q21.1-chr21q21.3 and the phenotypic diversity of Down’s syndrome (DS; trisomy 21). J. Nat. Sci. 2017, 3, e446. [Google Scholar]
- Bofill-De Ros, X.; Santos, M.; Vila-Casadesus, M.; Villanueva, E.; Andreu, N.; Dierssen, M.; Fillat, C. Genome-wide miR-155 and miR-802 target gene identification in the hippocampus of Ts65Dn Down syndrome mouse model by miRNA sponges. BMC Genom. 2015, 16, 907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Y.; Alexandrov, P.N.; Pogue, A.I.; Zhao, Y.; Bhattacharjee, S.; Lukiw, W.J. miRNA-155 upregulation and complement factor H deficits in Down’s syndrome. Neuroreport 2012, 23, 168–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, A.; Vezzoli, M.; Busatto, S.; Paolini, L.; Faranda, T.; Abeni, E.; Caracausi, M.; Antonaros, F.; Piovesan, A.; Locatelli, C.; et al. Analysis of a nanoparticleenriched fraction of plasma reveals miRNA candidates for Down syndrome pathogenesis. Int. J. Mol. Med. 2019, 43, 2303–2318. [Google Scholar] [CrossRef] [Green Version]
- Tili, E.; Mezache, L.; Michaille, J.J.; Amann, V.; Williams, J.; Vandiver, P.; Quinonez, M.; Fadda, P.; Mikhail, A.; Nuovo, G. microRNA 155 up regulation in the CNS is strongly correlated to Down’s syndrome dementia. Ann. Diagn. Pathol. 2018, 34, 103–109. [Google Scholar] [CrossRef]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 8973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsilioni, I.; Theoharides, T.C. Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1beta. J. Neuroinflamm. 2018, 15, 239. [Google Scholar] [CrossRef] [PubMed]
- Podvin, S.; Jones, A.; Liu, Q.; Aulston, B.; Ransom, L.; Ames, J.; Shen, G.; Lietz, C.B.; Jiang, Z.; O’Donoghue, A.J.; et al. Dysregulation of Exosome Cargo by Mutant Tau Expressed in Human-induced Pluripotent Stem Cell (iPSC) Neurons Revealed by Proteomics Analyses. Mol. Cell Proteom. 2020, 19, 1017–1034. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Xu, Y.; Deng, W.; Zhang, L.; Zhu, H.; Yu, P.; Qu, Y.; Zhao, W.; Han, Y.; Qin, C. Brain Derived Exosomes Are a Double-Edged Sword in Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13, 79. [Google Scholar] [CrossRef]
- Backes, C.; Meese, E.; Keller, A. Specific miRNA Disease Biomarkers in Blood, Serum and Plasma: Challenges and Prospects. Mol. Diagn. 2016, 20, 509–518. [Google Scholar] [CrossRef]
- Aoyagi, A.; Condello, C.; Stohr, J.; Yue, W.; Rivera, B.M.; Lee, J.C.; Woerman, A.L.; Halliday, G.; van Duinen, S.; Ingelsson, M.; et al. Abeta and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci. Transl. Med. 2019, 11, eaat8462. [Google Scholar] [CrossRef]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control NDEV | DS-AD NDEV | |
|---|---|---|
| CD81 (ng/mL) | 15.21 | 23.51 |
| Aβ42 (pg/mL) | <LOD | 31.98 |
| Tau (pg/mL) | 23.12 | 35.38 |
| p-Tau T231 (pg/mL) | 57.01 | 242.26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledreux, A.; Thomas, S.; Hamlett, E.D.; Trautman, C.; Gilmore, A.; Rickman Hager, E.; Paredes, D.A.; Margittai, M.; Fortea, J.; Granholm, A.-C. Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain. J. Clin. Med. 2021, 10, 3931. https://doi.org/10.3390/jcm10173931
Ledreux A, Thomas S, Hamlett ED, Trautman C, Gilmore A, Rickman Hager E, Paredes DA, Margittai M, Fortea J, Granholm A-C. Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain. Journal of Clinical Medicine. 2021; 10(17):3931. https://doi.org/10.3390/jcm10173931
Chicago/Turabian StyleLedreux, Aurélie, Sarah Thomas, Eric D. Hamlett, Camille Trautman, Anah Gilmore, Emily Rickman Hager, Daniel A. Paredes, Martin Margittai, Juan Fortea, and Ann-Charlotte Granholm. 2021. "Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain" Journal of Clinical Medicine 10, no. 17: 3931. https://doi.org/10.3390/jcm10173931
APA StyleLedreux, A., Thomas, S., Hamlett, E. D., Trautman, C., Gilmore, A., Rickman Hager, E., Paredes, D. A., Margittai, M., Fortea, J., & Granholm, A. -C. (2021). Small Neuron-Derived Extracellular Vesicles from Individuals with Down Syndrome Propagate Tau Pathology in the Wildtype Mouse Brain. Journal of Clinical Medicine, 10(17), 3931. https://doi.org/10.3390/jcm10173931