Abstract
Non-alcoholic steatohepatitis (NASH) is a chronic liver disease affecting up to 6.5% of the general population. There is no simple definition of NASH, and the molecular mechanism underlying disease pathogenesis remains elusive. Studies applying single omics technologies have enabled a better understanding of the molecular profiles associated with steatosis and hepatic inflammation—the commonly accepted histologic features for diagnosing NASH, as well as the discovery of novel candidate biomarkers. Multi-omics analysis holds great potential to uncover new insights into disease mechanism through integrating multiple layers of molecular information. Despite the technical and computational challenges associated with such efforts, a few pioneering studies have successfully applied multi-omics technologies to investigate NASH. Here, we review the most recent technological developments in mass spectrometry (MS)-based proteomics, metabolomics, and lipidomics. We summarize multi-omics studies and emerging omics biomarkers in NASH and highlight the biological insights gained through these integrated analyses.
1. Introduction
In the past decade, high-throughput omics technologies have revolutionized biomedical research [1]. Obtaining multiple layers of molecular measurements such as genomics, transcriptomics, proteomics, metabolomics, and lipidomics helps to systematically understand health and disease states, and may uncover new biological insights into disease mechanisms. NASH is a severe form of non-alcoholic fatty liver disease (NAFLD) which may progress to irreversible end-stage liver disease (cirrhosis). It is also associated with an increased risk of complications from cardiovascular disease and kidney disease [2]. However, diagnostics and therapeutics are limited. Currently, NASH can only be diagnosed by pathological evaluation of liver biopsy, and is defined by the presence of excessive fat deposition in the liver exceeding 5% of hepatocytes, hepatocyte ballooning, and lobular inflammation, with or without fibrosis [3]. The pathogenesis of NASH has not been fully elucidated [4]. A “two-hit” hypothesis has been proposed in which “liver steatosis”, the “first hit” increases the susceptibility to NASH through a “second hit” such as endoplasmic reticulum and oxidative stress [5,6]. There are no markers with sufficient sensitivity and accuracy for the clinical use of non-invasive diagnosis of NASH [7,8]. Genome-wide association studies (GWAS) have identified robust and reproducible loci that contribute to NAFLD pathogenesis and variability of prognosis, including the non-synonymous single nucleotide polymorphisms (SNPs) in PNPLA3 (phospholipase domain-containing 3), TM6SF2 (transmembrane 6 superfamily member 2), MBOAT7 (membrane-bound O-acyltransferase domain-containing protein 7), GCKR (glucokinase regulator), and HSD17B13 (17-beta hydroxysteroid dehydrogenase 13) [9]. While the heritability estimates of NAFLD range from 20–70% in population, family-based, or twin studies, the proportion of heritability explained by known risk variants is still a modest 10–20% [10,11,12]. In addition, GWAS alone does not suffice to elucidate the functional roles of the identified genetic variation in disease onset and progression [13].
Regulation of gene expression gives rise to different cell types as determined by their transcriptional states, and therefore represents a pivotal link between genetic structure and the molecular phenotype. Transcriptomics can quantify up to tens of thousands of transcripts in cells or tissues and has been included in many routine biological studies. Single-cell RNA sequencing has identified 20 discrete resident cell populations in human liver providing an in-depth map of the human hepatic immune microenvironment [14]. A recent study applied bulk RNA sequencing to a group of 206 NAFLD patients, and identified gene expression signatures associated with early stages and stepwise progression of the disease [15]. Integration with publicly available single-cell RNAseq data allowed the authors to further dissect the likely relative contribution of specific intrahepatic cell populations to NAFLD pathogenesis and progression. These results showed that changes in the transcriptome represent potential clinically relevant markers of disease progression [15].
While transcriptomics gives a rough estimate of the expression level of transcripts into proteins, proteomics confirms the presence of proteins and provides direct measurements of their quantity and modification status, so it is closer to disease phenotype. Therefore, the study of protein profiles (proteomics) is integral to many research fields including biomarker discovery, drug development, and elucidation of disease mechanisms [16,17]. Metabolomics and its sub-field lipidomics are the most downstream members of the omics family. Despite the rapid progress in the field, the overwhelming chemical complexity and diversity of small biomolecules still pose great challenges to identification and quantification strategies and downstream bioinformatics analysis. Nevertheless, lipidomics is a very important technology in the study of NAFLD. Several lipid classes have been linked to lipotoxicity and progression of the disease [18,19]. Finally, an increasing number of studies applying multi-omics technologies to generate “big data” are being performed to address the pathophysiology and diagnostics of NASH.
In this review, we focus on the technological aspects of mass spectrometry (MS)-based omics and the integrated application of omics in NASH research. In the first of three sections, we describe MS-based proteomics, metabolomics, and lipidomics technologies with a focus on state-of-the-art technical workflows. This is followed by highlights from recent proteomics studies and a systematic literature review describing metabolomics and lipidomics studies in NASH. Finally, we summarize the existing literature on emerging omics biomarkers and the application of multi-omics to NASH research.
2. State-of-the-Art Proteomics, Metabolomics, and Lipidomics Technologies
2.1. MS-Based Proteomics
While genome sequencing deciphers the blueprint of human life, which is mostly static, the human proteome is a highly dynamic entity in terms of both number of proteoforms, their copy numbers, and their spatiotemporal expression. On top of the approximately 20,000 human protein-coding genes, a single protein-coding gene can easily produce as many as 100 proteoforms, including products of alternative splicing, those containing single amino acid polymorphisms arising from non-synonymous SNPs, and those carrying post-translational modifications (PTMs) [20,21].
In MS-based proteomics, “bottom-up” (or “shotgun”) proteomics is the most widely used workflow, in which proteins are subjected to proteolytic cleavage, and the resulting peptides are analyzed by liquid chromatography coupled online to tandem mass spectrometry (LC-MS/MS) [22,23,24]. Peptide identification relies on tandem MS/MS spectra matching to a database containing in silico or empirically generated peptide fragmentation patterns. A similarity score will be calculated to assign peptide-spectrum match (PSM) typically with a false discovery rate (FDR) controlled below 1% by a “target-decoy” approach [25]. Not all peptides can be detected by MS due to differences in their physicochemical properties, abundance, and ionization efficiency typically leading to a median sequence coverage of around 30% in tissue proteomes [26]. Consequently, proteins indistinguishable from each other based on identified peptides are grouped to form a protein group. The major alternative workflow to “bottom-up” proteomics is “top-down” proteomics, in which intact proteins are introduced into and measured by LC-MS/MS without enzymatic digestion [27], in principle allowing different proteoforms derived from one protein-coding gene to be distinguished. However, experimental challenges render this approach so far not amenable to large-scale proteomics investigations. In contrast, state-of-the-art bottom-up proteomics routinely identifies more than 6000 protein groups in cells and tissues in single run analyses and more than 10,000 protein groups after fractionation [26,28,29]. Blood plasma has one of the most complex proteomes with a dynamic range of protein concentrations of more than 10 orders of magnitude with the top 22 proteins comprising already 99% of total protein mass [30,31]. Due to the high dynamic range of plasma proteome and limitations in sensitivity that mass spectrometers can currently reach, measuring all plasma proteins remains elusive. The human Plasma Proteome Database (PPD) contains more than 10,000 protein products corresponding to 3778 distinct protein-coding genes [30,32]. The largest human plasma proteome dataset generated in a single study to data contains over 5300 proteins by ‘super-depletion’, extensive fractionation, and isobaric labelling—corresponding to 5002 genes [33]. These deep plasma proteomes entail additional experimental steps such as peptide fractionation and depletion of high-abundant proteins. These approaches increase the overall analysis time per sample and introduce variability to the workflow and are thus not preferred for large-scale proteomics investigations in a clinical setting [17]. At the current state of the MS technology, cost and investment of time are still often prohibitive for such workflows, even if low abundant proteins could be detected. The throughput and proteome depth of a given study have to be balanced depending on the budget and scope of the study. Depending on the LC-MS/MS instruments and acquisition methods used, current high-throughput methods, potentially applicable in the clinics, enable routine analysis of 30–60 plasma samples per day without depletion or pre-fractionation with a depth of 300–500 protein groups in a single run [34,35,36].
2.2. Proteomics Platforms beyond MS
While high-throughput MS-based plasma proteomics workflow routinely quantifies hundreds of the top abundant proteins, non-MS-based platforms in principle offer the simultaneous detection of thousands of proteins in a plasma sample. These technologies include the SOMAscan assay [37,38] and the proximity extension assay (PEA) commercialized by Olink Biosciences. Both technologies rely on reagents binding to proteins of interest (chemically modified nucleotides in SOMAscan and oligonucleotide-labeled antibody-pairs in PEA) for the “identification”, and the amplification of reporter sequences by quantitative real-time PCR or DNA microarrays for the quantification [37,39]. These immunoaffinity-based platforms could serve as complementarity to MS-based proteomics for detecting low-abundant proteins that are difficult to detect by MS, such as the Olink Inflammation panel that targets 92 inflammation-related protein biomarkers. However, there are long-recognized limitations associated with antibodies and other binders such as nonspecific binding and cross-reactivity, particularly in a highly multiplexed setting. Besides, both SOMAscan and the PEA assay are optimized for body fluid samples, i.e., plasma and serum, and are not designed for binding sites with PTMs or peptide variants that impede the binding of reagents.
MS-based proteomics has the advantage of specifically discovering and quantifying proteins in an untargeted manner, and is clearly the most powerful platform for analyzing tissue proteomes, PTMs, protein-protein interactions, and protein variants. In the case of plasma to solve the dynamic range issue, a recent trend is to combine multiple platforms to cover a broader range of proteins taking advantages of the complementary strengths of both targeted and untargeted approaches [40,41].
2.3. MS-Based Metabolomics and Lipidomics
Metabolomics is the study of metabolites broadly defined as non-peptide molecules of less than 1.5 kDa [42]. Lipidomics, as a subset of metabolomics, is dedicated to lipid analysis with tailored extraction protocols, analytical methods, and data analysis strategies [43,44,45,46]. The main polar compound classes in the human metabolome comprise carbohydrates, ketones, amino and other organic acids, as well as biogenic amides, whereas the hydrophobic ones, namely lipids, are grouped into eight categories, namely fatty acyls, glycerolipids, glycerophospholipids, sphingolipids, saccharolipids, polyketides, sterol and prenol lipids [47] (Table 1). Among these small molecules, bile acids are of particular interest in NASH given their potent roles in mediating metabolic functions [48], as illustrated by the fact that several agonists of the bile acid receptor—Farnesoid X receptor (FXR) and its downstream target FGF19 are in phase I and II trials in treating NASH [49,50,51]. The structural diversity of the human metabolome poses a major challenge for analytical methods [52] resulting in various analytical approaches suited for detecting different classes of small molecules based on MS: LC-MS, gas chromatography mass spectrometry (GC-MS), imaging mass spectrometry, capillary electrophoresis–mass spectrometry, nuclear magnetic resonance, and Fourier transform infrared spectroscopy [53,54]. MS is the most commonly applied technology in metabolomics for the possibility of structural elucidation based on MS/MS spectra and metabolite annotation with higher confidence [55]. Compared with GC, where sample derivatization is often required, LC-MS based workflows are advantageous in clinical research for easier sample preparation. Hence, in the following section, we have chosen to focus on LC-MS-based workflows applied in metabolomics and lipidomics.

Table 1.
Overview of proteomics, LC-MS based metabolomics, and lipidomics platforms.
In a typical LC-MS-based metabolomics workflow, hydrophilic metabolites are extracted using solvents such as acetonitrile or methanol [56], followed by separation using reversed-phase LC with a C18 stationary phase or hydrophilic interaction LC (HILIC) prior to MS analysis [57]. In untargeted studies, mass analysis is typically performed via high-resolution, accurate mass MS instruments such as the Orbitrap or TOF analyzers [58,59,60]. Chromatographic peaks across samples are then detected and reported as a list of metabolic “features” for further statistical analysis. There are multiple commercial and freely accessible software packages for this, including MZmine [61], XCMS [62], MSDial [63], MetaboScape (Bruker Daltonics, Germany), and Compound Discoverer (Thermo, Germany). Annotation of detected features (metabolite identification) is done based on LC-MS related properties including accurate mass, retention time, tandem mass spectra, and recently ion mobility [64]. However, due to the enormous chemical diversity of possible isobaric and isomeric structures, the identification of metabolites and the elucidation of chemical structures remain challenging. To illustrate, searching the mass 181.07066 (glucose, M+H adduct) in the human metabolome database [65] even with a 5 ppm mass accuracy already yields 24 compounds, not including known unknowns (molecules that have previously been mass measured but not identified) as well as complete unknowns. Recent developments in bioinformatics aim at partially annotating unknown metabolites by comparing their tandem mass spectra to those of known ones existing in online databases [66,67,68,69,70].
Unlike hydrophilic metabolites, extraction of lipids from biological samples is typically done using highly apolar solvents, like chloroform and methyl tert-butyl ether (MTBE) following four most commonly used standardized methods [71,72,73,74,75]. MS analysis of lipid extracts is performed using either direct infusion (termed shotgun lipidomics) or in conjunction with LC [76]. In LC-MS-based approaches, reversed-phase analysis on C18 columns dominates, which separates lipid species of the same class based on the interaction of fatty acyl chains with the stationary phase. In contrast, HILIC mainly separates lipids by polar head groups. A recent trend is to integrate ion mobility spectrometry into conventional MS-based workflows [77], to separate ions in the gas phase by their size and shape, which can be advantageous in resolving isomers. We have recently demonstrated the benefits of trapped ion mobility spectrometry and a highly sensitive data acquisition method (PASEF) in generating comprehensive lipidomics profiles from a small sample amount equivalent to 10 µg of liver tissue per injection [64]. Feature detection in lipidomics is often performed using the same tools as the polar part of the metabolome, but lipid annotation is done using dedicated modules and separate software [78,79]. Despite the seemingly simple structure of lipids, annotation faces various challenges arising from the multitude of isomers due to the positioning of double bonds and acyl chains in the molecule. In addition, liver and plasma samples might also contain lipids of odd-chain fatty acids derived from food intake and bacterial products in the gastrointestinal tract [80].
3. Proteomics-Based Biomarker Discovery Studies in Liver Disease
Hundreds of proteomics-based biomarker discovery studies in liver disease have been reported during the past two decades. In a recent literature review, we observed a significant bias towards hepatocellular carcinoma (HCC) and viral hepatitis among all causes of liver diseases, with only a small fraction of studies focusing on NAFLD and alcohol-related liver disease (ALD) despite them being the most prevalent types of liver disease [87]. More than 200 different proteins potentially useful for the diagnosis, prognosis, and progression stratification in NAFLD have been reported, typically in the form of a list of dysregulated proteins [88]. However, these can be difficult to interpret for clinicians or researchers engaged in translational research. Only a few of these studies took a step further to demonstrate the predictive or discriminative power of proposed biomarkers by building machine learning-based classification models, often predicting only one type of pathological condition: fatty liver [89,90], and recently fibrosis [91]. In addition, currently proposed candidate biomarkers suffered from low reproducibility and robustness, demonstrated by only one overlapping protein—MET (hepatocyte growth factor receptor) in the proposed protein marker panels for fatty liver in the two above-mentioned studies using immunoaffinity-based proteomics platforms. Furthermore, simply diagnosing fatty liver does not help clinical decisions, which are more concerned with liver fibrosis, the strongest predictor of liver- and all cause-related mortality as well as hepatic inflammation, which reflects disease activity [92]. In a recent study, a 12-protein panel was identified using the SomaScan proteomics platform which can distinguish between fibrosis stages F0–1 and F2–4 in patients with NAFLD with an area under the Receiver Operating Characteristics curve (AUROC) of 0.74 [91].
Recent progress in MS-based proteomics has enabled the generation of large datasets in clinical studies, accompanied by increasingly reproducible results. In an early effort, we identified polymeric immunoglobulin receptor (PIGR) as a predictor of NAFLD independent of insulin resistance [36], and this association between PIGR and NAFLD was subsequently reproduced in other studies [35,93,94]. Even though the focus of this review is NAFLD, ALD is indistinguishable under the microscope in terms of histological features, and hence might share common biomarkers. In a more recent effort, we acquired plasma proteomes from close to 600 individuals of biopsy-verified ALD and healthy controls, as well as 79 liver proteomes from the disease group [35]. Among the major findings, we identified proteomic marker panels to predict significant liver fibrosis (AUROC = 0.88), mild inflammation (AUROC = 0.83), and any presence of steatosis (AUROC = 0.89) with superior or comparable performance compared to existing best-in-class clinical tests including the FibroScan, the M30 apoptosis marker for hepatic inflammation, and the CAP value for liver steatosis. By integrating proteome changes in paired liver- and plasma samples, we could attribute the tissue origins of many of the proposed candidate markers. Comparing with the previous NAFLD study, three proteins PIGR, ALDOB, and LGALS3BP were common and robust markers for NAFLD and ALD. Given a NAFLD study of equivalent size and patient heterogeneity, it is likely to identify more circulating markers common to NAFLD and ALD. Recently, PIGR was also reported to be upregulated in patients with COVID-19 infection [95], possibly indicating it might not be specific to liver disease but reflect a general inflammation process. In any case, based on current results, PIGR is an indicator of hepatic inflammation and liver fibrosis in the context of liver disease. Importantly, proteomics-based biomarker discovery allows the identification of not only one single protein but rather panels of proteins, which collectively reflect the complex nature of the disease pathology and the need to study it from a systems biology perspective [35].
4. Metabolomics-Based Biomarker Discovery Studies in NASH
To provide an overview of recent metabolomics studies in NASH, we systematically searched for publications in the PubMed database using the logic terms “(nonalcoholic steatohepatitis OR NASH OR non-alcoholic fatty liver disease OR NAFLD) AND (lipidomics OR metabolomics) AND (human OR clinical)” for the period from 1 September 2015 to 1 September 2020. In this review, we only considered original research articles, which use MS and human samples. High complexity of the liver metabolome has opened up various MS applications in biomarker discovery (Table 2), ranging from polar metabolites [96] to lipids [97] using both targeted [98] and increasingly popular untargeted approaches [99]. Most of the studies shown in Table 2 reported perturbations in triglycerides, amino acids, fatty acids, and basic mitochondrial energy metabolism in NASH/NAFLD. Due to the diverse changes associated with NAFLD/NASH across many classes of lipids and metabolites, there is no clear consensus among the studies on candidate biomarkers or biochemical pathways (Table 2). This is potentially due to the large inter-individual variations in the metabolome and its extremely dynamic nature. Having a separate validation cohort for the biological confirmation of newly identified biomarker signatures might help to avoid misinterpretation of any study outcome and achieve more reproducible findings. Making data publicly available can further promote reproducible and transparent research. Surprisingly, our review shows that only three out of the 25 reviewed publications validated their findings in a separate study [100,101,102]. Moreover, none of the 25 studies released data in a public repository for future meta-analyses, although an initiative to standardize the reporting of metabolomics studies has been formed years ago [103,104]. Nine of the 25 studies not only proposed potential biomarkers but also evaluated the classification performance. These proposed marker candidates are summarized in Table 3 together with other omics markers. In brief, sample sizes range from 31 to 1479 with five studies having a sample size of below 100. Only five studies validated the marker performance in a validation cohort, with sample sizes ranging from 22 to 192. Most of these studies focus on predicting NASH in NAFLD patients, with a few exceptions, which predict significant or advanced fibrosis in patients with NASH, or distinguish between NAFLD and healthy individuals [100,102,105,106]. Based on these studies, circulating metabolome has good predictive power in identifying NASH and fibrosis in patients with NAFLD, as well as distinguishing between patients with NAFLD and healthy individuals. With a logistic regression model based on a biomarker panel consisted of eight lipids, one amino acid, and one carbohydrate, the AUROC for identifying advanced fibrosis (F3–4) in NAFLD was 0.94 in the discovery cohort (n = 156) and 0.84 in the validation cohort (n = 142) [100]. In another study of a smaller cohort (n = 31), an AUROC of 1.0 was achieved in predicting significant fibrosis (F2–4) with a support vector machine based on a marker panel of 10 lipids including diglycerides, triglycerides, and (lyso)phosphatidylcholines [105]. However, a validation cohort was not provided. Using a panel of 11 triglycerides or a combination of 11 metabolite features and three clinical markers, an AUROC of 0.9 and 0.94 was achieved respectively in identifying patients with NAFLD against healthy individuals [102,106]. Similarly, modest to high performance was achieved in predicting NASH in patients with NAFLD with AUROCs ranging between 0.65 and 0.95 (Table 3). Agreements of the AUROC between discovery and validation cohorts are generally good, with extremes differing as much as 0.16 (worse in validation) [102]. Apart from the highly dynamic nature of the human circulating metabolome, a few additional factors may contribute to such huge discrepancy in model performance between discovery and validation cohorts including differences in the distribution of disease severity, over-fitting in model training, or underpowered study design.
Table 2.
Overview of MS-based metabolomics in NASH/NAFLD human studies.
Table 3.
Emerging omics markers and their classification performance for diagnosing NAFLD/NASH.
Similar to metabolomics, we retrieved publications from PubMed database using the logic terms “(nonalcoholic steatohepatitis OR NASH OR non-alcoholic fatty liver disease OR NAFLD) AND (multiomics OR multi-omic)”, for the period from 1 September 2015 to 1 September 2020. This search strategy generated 27 records. We only considered articles that were not reviews or conference proceedings. The PubMed query did not retrieve three other relevant works, which we added manually. In total, this resulted in 14 papers meeting our criteria (Figure 1 and Table 4). We were first surprised by the small number of studies that have applied multi-omics techniques in this field so far. Although irrelevant to this review, replacing the keyword of “NASH” to “liver disease”, the search query resulted in 114 records, with a large proportion of studies focusing on hepatocellular carcinoma and other types of liver cancer. These search results implied limited resources of multi-omics datasets that have been generated on the topic of NASH, and a study bias towards liver cancer among all liver diseases, which is in concordance with a recent review on plasma proteomics efforts in liver disease [87]. Below, we describe the omics data types, research aims, experimental design, data integrative strategies, and study outcomes of the selected papers.
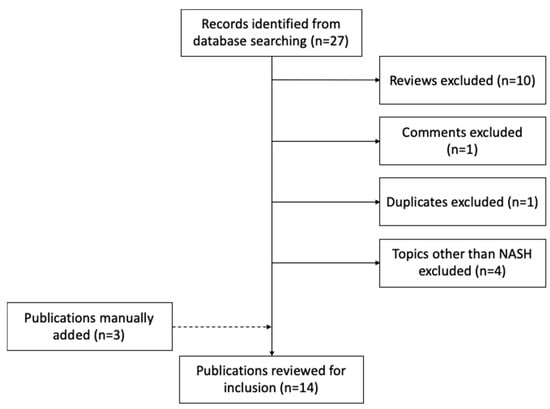
Figure 1.
Flow chart of studies identified, excluded, and included.
Table 4.
Overview of multi-omics studies in NASH/NAFLD.
4.1. Characteristics of Studies
Among these 14 studies, six characterized a specific biological or disease model using multi-omics datasets. For instance, a systemic approach was used to characterize the molecular alterations of a carbohydrate-restricted diet on hepatic steatosis in humans [124], and to describe the molecular profiles of a diet-induced obese model of NASH [125]. Only three studies focused on finding biomarkers or identifying discriminative molecular signatures for predicting fatty liver disease using multi-omics data [89,90,94]. These studies performed omics technologies on human (36%), mouse, or rat (43%) or a combination of both (21%) (Figure 2a). In terms of sample types, most of the studies used liver biopsies followed by blood plasma/serum, fecal samples, and adipose tissue of human and rodent origin (Figure 2b).
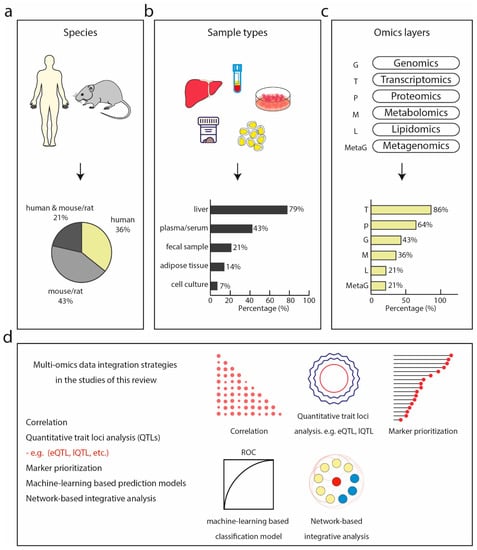
Figure 2.
Multi-omics studies in NASH. (a) Species of which bio-specimens were used in the surveyed studies including human, model organisms such as mouse and rat, or a combination of both. (b) Sample types collected and analyzed by omics technologies in the surveyed studies, dominated by liver, followed by plasma or serum, fecal samples, adipose tissue, and cell culture. (c) Omics technologies used in the surveyed studies with transcriptomics being the most commonly used, followed by proteomics, genomics, metagenomics, lipidomics, and metagenomics. (d) Multi-omics integration strategies used in the surveyed studies.
Transcriptomics was the most frequently performed (86% of all studies), followed by proteomics (64%) and genotyping (43%) (Figure 2c). Metabolome, lipidome, and metagenome were the least commonly generated data types, accounting for only 36%, 21%, and 21%, respectively. Transcriptomics and proteomics are most frequently combined. This could reflect to some extent the maturity, throughput, and accessibility of these technologies to non-specialized researchers. The majority of these studies generated new omics data along with the publications, however, only half of them made the data publicly accessible. The inaccessibility of publicly available datasets in turn hinders in silico-only studies. Most of the RNA sequencing data were made publicly available at the NCBI Gene Expression Omnibus and the NCBI Sequence Read Archive (SRA) database. Among the nine studies that included proteomics data, five used MS-based proteomics with the remaining adopting antibody-based approaches. Despite the growing consensus in the proteomics community about making mass spectrometry raw data accessible and reusable by uploading to a public database like PRIDE [135], only one study [94] did so (project identifier: PXD014751). In line with what we found in our above-described metabolomics review, only one study [126] deposited metabolomics data at the MetaboLights database (https://www.ebi.ac.uk/metabolights/), a database for metabolomics experiments maintained by the European Bioinformatics Institute (EMBO EBI). One study [130] deposited lipidomics mass spectrometry data at the Chorus project (http://chorusproject.org).
4.2. Overview of Data Integration Strategies
One of the advantages of applying multi-omics technologies to the same biological system is to understand the flow of information underlying disease and interpret the data in a holistic way in the context of biological networks and molecular interactions. Currently, omics data integration methods generally fall into two categories: multi-staged analysis and meta-dimensional analysis [136]. The difference between these two approaches is that multi-staged analysis performs data integration in a stepwise manner, adding one additional omics layer at a time, whereas meta-dimensional analysis attempts to incorporate and analyze all the types of data simultaneously. A systematic review of such existing tools can be found elsewhere [137]. In the surveyed literature, data integration was performed at different stages, predominantly at data analysis (data level, Table 4), followed by statistical and pathway data integration (result level, Table 4). Among those that perform integration at data level, various bioinformatics techniques were used, including machine learning-based approaches [89,90,134], correlation between two data types [129,130,134], quantitative trait loci (QTL) analysis [130,132], and network-based association analysis [132,133] for integrating more than one dataset, and weighted gene co-expression network analysis (WGCNA) [130] on a single layer of omics data (Figure 2d). Functional enrichment analysis including gene set enrichment analysis (GSEA) for KEGG pathways and GO terms were commonly employed in studies that integrate data at the level of statistical and bioinformatics results [125,127,128] (Supplemental Table S1). In one of them, the authors performed liver proteomics and metabolomics analysis to investigate the molecular mechanism underlying the Roundup pesticide in inducing liver pathology using a rat model [128]. By performing differential expression analysis followed by functional annotation using pathway analysis tools, the authors identified proteome changes associated with lipid detoxifying metabolic processes indicating lipid peroxidation, oxidative stress, and hepatocyte injury, all NASH-like pathological features. This association with a NASH-like phenotype was further supported in the metabolome profile by an increase in metabolites of oxidative stress and fibrosis markers.
4.3. Multi-Omics Classifiers and Discriminative Disease Signatures
When the aim is to select predictive features for disease, machine learning approaches can treat multi-omics variables equally, also considering interaction between variables across omics layers. Three studies performed model-based integration at the data level to identify discriminative omics signatures for predicting disease phenotype [89,90,94]. Baseline data from the deep phenotyped IMI DIRECT cohorts (n = 1514) were used to build machine learning models for predicting NAFLD [89]. With a selected set of clinical and omics variables, a random forest machine learning model predicts NAFLD with an AUROC of 0.84, higher than those using only clinical data or any other omics data alone. Interestingly, when examining the predictive ability of each omics dataset as input variables alone, proteomic markers yielded the highest predictive accuracy surpassing genetic-, blood transcriptomics-, and metabolomics data. The proteomics data generated in this study derived from a combination of various immunoassays that target proteins with known associations to disease. Whether the use of an unbiased proteomics technology, i.e., MS-based proteomics, affects the predictive accuracy requires further investigation. In another biomarker discovery study, a multi-component classifier for NAFLD was developed, based on genotyping, serum proteomics, and clinical data such as plasma glucose level, HDL, and ALT [90]. The authors assessed the performance of classifiers based on each data domain alone and found that proteomics achieved the highest AUROC of 0.913, followed by phenomics data (0.886) and PNPLA3 genotyping data (0.596). Combining all markers selected from each individual data domain achieved an AUROC of 0.935. Similarly, in a biomarker discovery pre-clinical study, liver transcriptomics and proteomics as well as plasma proteomics were performed on a rat model of NASH aiming to characterize the molecular pathophysiology of NASH and to identify new plasma biomarkers [94]. By collecting molecular signals associated with NASH pathogenesis, the authors developed a multi-dimensional ranking approach integrating multi-omics data with liver histology characterization and prior knowledge and uncovered known as well as novel marker candidates of NASH and fibrosis. This study demonstrated that the integration of liver transcriptomics with liver- and plasma proteomics captured the translation of molecular changes from the diseased liver at the RNA level to the changes of liver and plasma protein level, and increased the biological resolution of discovered potential non-invasive biomarkers. Of the above-mentioned studies, only the one that utilized the IMI DIRECT data performed external validation using the UK biobank cohort on selected prediction models that were built on widely available clinical parameters.
5. Conclusions and Prospects
MS-based omics technologies are powerful tools to study human health and disease, and have a great potential to revolutionize tomorrow’s clinical laboratory diagnosis. Despite the extremely low translation rate of basic scientific findings into clinical applications in the early efforts, we are starting to see more reproducible and convincing results generated across clinical cohorts by independent research groups, especially in biomarker discovery studies in liver disease using MS-based proteomics. As clinical proteomics is increasingly capable of large-scale analysis of patient samples, machine learning-based approaches are emerging in large clinical studies to demonstrate the predictive power of newly identified composite marker panels. Looking forward, the FDA has already cleared a few MS-based devices for clinical use. However, as of today, no LC-MS-based diagnostic test that measures proteins or peptides has been approved. Apart from biological and clinical validation, a robust and quantitative proteomics assay needs to be established and validated across hospital sites and instruments to be used in the clinic.
Existing clinical metabolomics and lipidomics studies in NASH have unveiled a broad range of changes in multiple classes of metabolites and lipids. A few studies have also identified potential biomarker panels for detecting different stages of fibrosis and NASH in NALFD. However, collectively they do not converge in terms of the core dysregulated metabolic pathways or potential biomarkers. As we have argued in the review, a well-designed clinical study including the use of a validation cohort, standardization of the experimental pipeline, and the potential release of the research data can help generate reproducible and robust results, further unlocking the real power of clinical metabolomics and lipidomics. Several pioneering studies have already integrated multi-omics data types generated on the same cohorts to build classifiers for detecting NAFLD, including genotyping, immunoaffinity-based proteomics, and MS-based metabolomics. Despite the minimal overlap among the proposed biomarker panels in previous literature, these newer studies clearly demonstrate the advantages of model performance when integrating multiple layers of omics information compared with using single layers of omics data alone.
A common issue of omics-based biomarker discovery is the lack of classification performance of the proposed biomarkers, and the lack of verification in independent cohorts. Good practice in machine learning is necessary for training reliable, repeatable, and reproducible models [138]. In general, external validation in independent cohorts is always required to test the generalization ability of a learned model. From the surveyed literature, we have observed that there is a moderate to good agreement in the predictive power of candidate markers between discovery and validation cohorts. However, some studies also show great discrepancies. As we inferred, this may be due to differences in disease severity distribution, poor or insufficiently robust technical workflows for generating omics data, overfitting during model training, or underpowered study design. Considering these elements during study design will increase the success rate in future biomarker discovery studies and the subsequent implementation in clinical practice. Depending on the performance evaluation strategy and the disease severity distribution of the study population, it may be difficult to compare model performance across studies. This should also be taken into consideration when evaluating performance of emerging markers, especially across platforms. As more and more data are generated in clinical studies of NASH/NAFLD, it is promising to develop a powerful composite marker panel based on omics to detect disease. In addition to improving predictive power, compared with traditional markers that usually focus on a single aspect of the disease, multi-omics composite biomarker panels may also capture more biological complexity of disease pathogenesis and progression. However, if omics-based marker panels only provide marginal gain in terms of diagnostic performance compared to the best performing omics data type, practically it may be preferred to develop a diagnostic test based on a single technology. We believe that future research should focus on identifying diagnostic markers that can detect early stages of fibrosis and NASH in high-risk populations, such as individuals with obesity or type 2 diabetes. In addition, only a small percentage of patients progress from simple steatosis to NASH. Such predictive markers of can also benefit the clinical management of disease progression. We predict that prospective longitudinal studies to identify omics-based predictors of disease progression and therapeutic response will help to provide an alternative to liver biopsy, thereby avoiding unnecessary invasive testing and expediting drug development. In addition, the integration of omics datasets through powerful computational methods will help infer causality and reveal new insights into disease mechanisms. Finally, image based spatial omics provides unique opportunities to study the molecular profile of tissue sections at the level of single cells and organelles. In spatial metabolomics in particular, it has become possible to localize metabolites, lipids, and drugs in tissue sections through imaging mass spectrometry [139]. Although spatial proteomics and metabolomics are emerging fields, they will be a very valuable addition to research in liver diseases.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jcm10204673/s1, Table S1: Metadata of the reviewed literature.
Funding
The research described was funded by The Novo Nordisk Foundation for the Clinical Proteomics group (grant NNF15CC0001), the Copenhagen Bioscience PhD Program (grant NNF16CC0020906), and the Challenge Programme MicrobLiver (grant NNF15OC0016692). NJWA was supported by an Novo Nordisk Foundation Excellence Emerging Investigator Grant—Endocrinology and Metabolism (application No. NNF19OC0055001). Furthermore, LN and SR were supported by the Novo Nordisk Foundation (grant NNF14CC0001).
Acknowledgments
We thank all members of the Clinical Proteomics group at Novo Nordisk Foundation Center for Protein Research and the Proteomics and Signal Transduction Group (Max Planck Institute).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Targher, G. Increased risk of cardiovascular disease and chronic kidney disease in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 372–381. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Gentile, C.L.; Pagliassotti, M.J. The role of fatty acids in the development and progression of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2008, 19, 567–576. [Google Scholar] [CrossRef]
- Long, M.T.; Gandhi, S.; Loomba, R. Advances in non-invasive biomarkers for the diagnosis and monitoring of non-alcoholic fatty liver disease. Metab. Clin. Exp. 2020, 111, 154259. [Google Scholar] [CrossRef]
- Lambrecht, J.; Tacke, F. Controversies and Opportunities in the Use of Inflammatory Markers for Diagnosis or Risk Prediction in Fatty Liver Disease. Front. Immunol. 2020, 11, 634409. [Google Scholar] [CrossRef]
- Trépo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Di Costanzo, A.; Belardinilli, F.; Bailetti, D.; Sponziello, M.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Pastori, D.; Ceci, F.; Montali, A.; et al. Evaluation of Polygenic Determinants of Non-Alcoholic Fatty Liver Disease (NAFLD) By a Candidate Genes Resequencing Strategy. Sci. Rep. 2018, 8, 3702. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic contributions to NAFLD: Leveraging shared genetics to uncover systems biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Genetic predisposition in nonalcoholic fatty liver disease. Clin. Mol. Hepatol 2017, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Aprile, M.; Esposito, R.; Ciccodicola, A. RNA-Seq and human complex diseases: Recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Govaere, O.; Cockell, S.; Tiniakos, D.; Queen, R.; Younes, R.; Vacca, M.; Alexander, L.; Ravaioli, F.; Palmer, J.; Petta, S.; et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020, 12, eaba4448. [Google Scholar] [CrossRef]
- Frantzi, M.; Latosinska, A.; Mischak, H. Proteomics in Drug Development: The Dawn of a New Era? Proteom. Clin. Appl 2019, 13, e1800087. [Google Scholar] [CrossRef] [PubMed]
- Geyer, P.E.; Holdt, L.M.; Teupser, D.; Mann, M. Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol. 2017, 13, 942. [Google Scholar] [CrossRef] [PubMed]
- Kartsoli, S.; Kostara, C.E.; Tsimihodimos, V.; Bairaktari, E.T.; Christodoulou, D.K. Lipidomics in non-alcoholic fatty liver disease. World J. Hepatol. 2020, 12, 436–450. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Pierantonelli, I.; Torquato, P.; Marinelli, R.; Ferreri, C.; Chatgilialoglu, C.; Bartolini, D.; Galli, F. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 2019, 144, 293–309. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd; Speicher, S.; Griffin, P.R.; Hunkapiller, T. Peptide mass maps: A highly informative approach to protein identification. Anal. Biochem. 1993, 214, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.H.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- Kelleher, N.L.; Lin, H.Y.; Valaskovic, G.A.; Aaserud, D.J.; Fridriksson, E.K.; McLafferty, F.W. Top Down versus Bottom Up Protein Characterization by Tandem High-Resolution Mass Spectrometry. J. Am. Chem. Soc. 1999, 121, 806–812. [Google Scholar] [CrossRef]
- Meier, F.; Geyer, P.E.; Virreira Winter, S.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Bekker-Jensen, D.B.; Kelstrup, C.D.; Batth, T.S.; Larsen, S.C.; Haldrup, C.; Bramsen, J.B.; Sorensen, K.D.; Hoyer, S.; Orntoft, T.F.; Andersen, C.L.; et al. An Optimized Shotgun Strategy for the Rapid Generation of Comprehensive Human Proteomes. Cell Syst. 2017, 4, 587–599.e584. [Google Scholar] [CrossRef]
- Nanjappa, V.; Thomas, J.K.; Marimuthu, A.; Muthusamy, B.; Radhakrishnan, A.; Sharma, R.; Ahmad Khan, A.; Balakrishnan, L.; Sahasrabuddhe, N.A.; Kumar, S.; et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014, 42, D959–D965. [Google Scholar] [CrossRef]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. MCP 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Muthusamy, B.; Hanumanthu, G.; Suresh, S.; Rekha, B.; Srinivas, D.; Karthick, L.; Vrushabendra, B.M.; Sharma, S.; Mishra, G.; Chatterjee, P. Plasma Proteome Database as a resource for proteomics research. Proteomics 2005, 5, 3531–3536. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Gillette, M.A.; Mertins, P.; Clauser, K.R.; Mani, D.R.; Kuhn, E.W.; Farrell, L.A.; Gerszten, R.E.; Carr, S.A. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Mol. Cell. Proteom. 2015, 14, 2375–2393. [Google Scholar] [CrossRef]
- Bruderer, R.; Muntel, J.; Müller, S.; Bernhardt, O.M.; Gandhi, T.; Cominetti, O.; Macron, C.; Carayol, J.; Rinner, O.; Astrup, A.; et al. Analysis of 1508 Plasma Samples by Capillary-Flow Data-Independent Acquisition Profiles Proteomics of Weight Loss and Maintenance. Mol. Cell. Proteom. MCP 2019, 18, 1242–1254. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Thiele, M.; Geyer, P.E.; Rasmussen, D.N.; Webel, H.E.; Santos, A.; Gupta, R.; Meier, F.; Strauss, M.; Kjaergaard, M.; et al. A paired liver biopsy and plasma proteomics study reveals circulating biomarkers for alcohol-related liver disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef] [PubMed]
- Hensley, P. SOMAmers and SOMAscan—A Protein Biomarker Discovery Platform for Rapid Analysis of Sample Collections From Bench Top to the Clinic. J. Biomol. Tech. JBT 2013, 24, S5. [Google Scholar]
- Billing, A.M.; Ben Hamidane, H.; Bhagwat, A.M.; Cotton, R.J.; Dib, S.S.; Kumar, P.; Hayat, S.; Goswami, N.; Suhre, K.; Rafii, A.; et al. Complementarity of SOMAscan to LC-MS/MS and RNA-seq for quantitative profiling of human embryonic and mesenchymal stem cells. J. Proteom. 2017, 150, 86–97. [Google Scholar] [CrossRef]
- Berggrund, M.; Ekman, D.; Gustavsson, I.; Sundfeldt, K.; Olovsson, M.; Enroth, S.; Gyllensten, U. Protein Detection Using the Multiplexed Proximity Extension Assay (PEA) from Plasma and Vaginal Fluid Applied to the Indicating FTA Elute Micro Card™. J. Circ. Biomark. 2016, 5, 9. [Google Scholar] [CrossRef]
- Petrera, A.; von Toerne, C.; Behler, J.; Huth, C.; Thorand, B.; Hilgendorff, A.; Hauck, S.M. Multiplatform Approach for Plasma Proteomics: Complementarity of Olink Proximity Extension Assay Technology to Mass Spectrometry-Based Protein Profiling. J. Proteome Res. 2021, 20, 751–762. [Google Scholar] [CrossRef]
- Finkernagel, F.; Reinartz, S.; Schuldner, M.; Malz, A.; Jansen, J.M.; Wagner, U.; Worzfeld, T.; Graumann, J.; von Strandmann, E.P.; Müller, R. Dual-platform affinity proteomics identifies links between the recurrence of ovarian carcinoma and proteins released into the tumor microenvironment. Theranostics 2019, 9, 6601–6617. [Google Scholar]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- Züllig, T.; Trötzmüller, M.; Köfeler, H.C. Lipidomics from sample preparation to data analysis: A primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Simons, K. Lipidomics: Coming to grips with lipid diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. [Google Scholar] [CrossRef]
- Wenk, M.R. Lipidomics: New tools and applications. Cell 2010, 143, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Han, X. Lipidomics for studying metabolism. Nat. Rev. Endocrinol. 2016, 12, 668–679. [Google Scholar] [CrossRef]
- Pradas, I.; Huynh, K.; Cabré, R.; Ayala, V.; Meikle, P.J.; Jové, M.; Pamplona, R. Lipidomics Reveals a Tissue-Specific Fingerprint. Front. Physiol. 2018, 9, 1165. [Google Scholar] [CrossRef]
- Molinaro, A.; Wahlström, A.; Marschall, H.U. Role of Bile Acids in Metabolic Control. Trends Endocrinol. Metab. TEM 2018, 29, 31–41. [Google Scholar] [CrossRef]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients with Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231.e211. [Google Scholar] [CrossRef]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients with Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Rampler, E.; Abiead, Y.E.; Schoeny, H.; Rusz, M.; Hildebrand, F.; Fitz, V.; Koellensperger, G. Recurrent Topics in Mass Spectrometry-Based Metabolomics and Lipidomics—Standardization, Coverage, and Throughput. Anal. Chem. 2021, 93, 519–545. [Google Scholar] [CrossRef]
- Dunn, W.B.; Bailey, N.J.C.; Johnson, H.E. Measuring the metabolome: Current analytical technologies. Analyst 2005, 130, 606–625. [Google Scholar] [CrossRef]
- Domenick, T.M.; Gill, E.L.; Vedam-Mai, V.; Yost, R.A. Mass Spectrometry-Based Cellular Metabolomics: Current Approaches, Applications, and Future Directions. Anal. Chem. 2021, 93, 546–566. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Ellis, D.I. Metabolomics: Current analytical platforms and methodologies. TrAC Trends Anal. Chem. 2005, 24, 285–294. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- García-Cañaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. A comprehensive untargeted metabonomic analysis of human steatotic liver tissue by RP and HILIC chromatography coupled to mass spectrometry reveals important metabolic alterations. J. Proteome Res. 2011, 10, 4825–4834. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.J.; Schultz, A.W.; Wang, J.; Johnson, C.H.; Yannone, S.M.; Patti, G.J.; Siuzdak, G. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013, 8, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab Dis 2018, 41, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L.; et al. Systems biology guided by XCMS Online metabolomics. Nat. Methods 2017, 14, 461–462. [Google Scholar] [CrossRef]
- Tsugawa, H.; Cajka, T.; Kind, T.; Ma, Y.; Higgins, B.; Ikeda, K.; Kanazawa, M.; VanderGheynst, J.; Fiehn, O.; Arita, M. MS-DIAL: Data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [Google Scholar] [CrossRef]
- Vasilopoulou, C.G.; Sulek, K.; Brunner, A.-D.; Meitei, N.S.; Schweiger-Hufnagel, U.; Meyer, S.W.; Barsch, A.; Mann, M.; Meier, F. Trapped ion mobility spectrometry and PASEF enable in-depth lipidomics from minimal sample amounts. Nat. Commun. 2020, 11, 331. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Nothias, L.-F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra. Nat. Biotechnol. 2021, 39, 462–471. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Löfgren, L.; Forsberg, G.B.; Ståhlman, M. The BUME method: A new rapid and simple chloroform-free method for total lipid extraction of animal tissue. Sci. Rep. 2016, 6, 27688. [Google Scholar] [CrossRef]
- Löfgren, L.; Ståhlman, M.; Forsberg, G.B.; Saarinen, S.; Nilsson, R.; Hansson, G.I. The BUME method: A novel automated chloroform-free 96-well total lipid extraction method for blood plasma. J. Lipid Res. 2012, 53, 1690–1700. [Google Scholar] [CrossRef]
- Matyash, V.; Liebisch, G.; Kurzchalia, T.V.; Shevchenko, A.; Schwudke, D. Lipid extraction by methyl-tert-butyl ether for high-throughput lipidomics. J. Lipid Res. 2008, 49, 1137–1146. [Google Scholar] [CrossRef]
- Holčapek, M.; Liebisch, G.; Ekroos, K. Lipidomic Analysis. Anal. Chem. 2018, 90, 4249–4257. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Smith, A.J.; Astarita, G. Ion mobility mass spectrometry in the omics era: Challenges and opportunities for metabolomics and lipidomics. Mass Spectrom. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Arita, M. The importance of bioinformatics for connecting data-driven lipidomics and biological insights. Biochim. Et Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 762–765. [Google Scholar] [CrossRef]
- Van Meer, G.; De Kroon, A.I.P.M. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef]
- Doll, S.; Dreßen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.-A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef]
- Chaleckis, R.; Meister, I.; Zhang, P.; Wheelock, C.E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Curr. Opin. Biotechnol. 2019, 55, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Telu, K.H.; Yan, X.; Wallace, W.E.; Stein, S.E.; Simón-Manso, Y. Analysis of human plasma metabolites across different liquid chromatography/mass spectrometry platforms: Cross-platform transferable chemical signatures. Rapid Commun. Mass Spectrom. RCM 2016, 30, 581–593. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, S.H.; Kim, J.H.; Hwang, S.; Yoo, H.J. Understanding Metabolomics in Biomedical Research. Endocrinol. Metab. 2016, 31, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.A.; Heckert, A.; Ulmer, C.Z.; Jones, C.M.; Koelmel, J.P.; Abdullah, L.; Ahonen, L.; Alnouti, Y.; Armando, A.M.; Asara, J.M.; et al. Harmonizing lipidomics: NIST interlaboratory comparison exercise for lipidomics using SRM 1950-Metabolites in Frozen Human Plasma. J. Lipid Res. 2017, 58, 2275–2288. [Google Scholar] [CrossRef]
- Thompson, J.W.; Adams, K.J.; Adamski, J.; Asad, Y.; Borts, D.; Bowden, J.A.; Byram, G.; Dang, V.; Dunn, W.B.; Fernandez, F.; et al. International Ring Trial of a High Resolution Targeted Metabolomics and Lipidomics Platform for Serum and Plasma Analysis. Anal. Chem. 2019, 91, 14407–14416. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Geyer, P.E.; Mann, M. Proteomics in the Study of Liver Diseases. In The Human Gut-Liver-Axis in Health and Disease; Krag, A., Hansen, T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 165–193. [Google Scholar] [CrossRef]
- Ladaru, A.; Balanescu, P.; Stan, M.; Codreanu, I.; Anca, I.A. Candidate proteomic biomarkers for non-alcoholic fatty liver disease (steatosis and non-alcoholic steatohepatitis) discovered with mass-spectrometry: A systematic review. Biomark. Biochem. Indic. Expo. Response Susceptibility Chem. 2016, 21, 102–114. [Google Scholar] [CrossRef]
- Atabaki-Pasdar, N.; Ohlsson, M.; Viñuela, A.; Frau, F.; Pomares-Millan, H.; Haid, M.; Jones, A.G.; Thomas, E.L.; Koivula, R.W.; Kurbasic, A.; et al. Predicting and elucidating the etiology of fatty liver disease: A machine learning modeling and validation study in the IMI DIRECT cohorts. PLoS Med. 2020, 17, e1003149. [Google Scholar] [CrossRef]
- Wood, G.C.; Chu, X.; Argyropoulos, G.; Benotti, P.; Rolston, D.; Mirshahi, T.; Petrick, A.; Gabrielson, J.; Carey, D.J.; DiStefano, J.K.; et al. A multi-component classifier for nonalcoholic fatty liver disease (NAFLD) based on genomic, proteomic, and phenomic data domains. Sci. Rep. 2017, 7, 43238. [Google Scholar] [CrossRef]
- Luo, Y.; Wadhawan, S.; Greenfield, A.; Decato, B.E.; Oseini, A.M.; Collen, R.; Shevell, D.E.; Thompson, J.; Jarai, G.; Charles, E.D.; et al. SOMAscan Proteomics Identifies Serum Biomarkers Associated with Liver Fibrosis in Patients With NASH. Hepatol. Commun. 2021, 5, 760–773. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Janech, M.G.; Sobolesky, P.M.; Bland, A.M.; Samsuddin, S.; Alazawi, W.; Syn, W.K. Proteomic screening of plasma identifies potential noninvasive biomarkers associated with significant/advanced fibrosis in patients with nonalcoholic fatty liver disease. Biosci. Rep. 2020, 40, BSR20190395. [Google Scholar] [CrossRef] [PubMed]
- Veyel, D.; Wenger, K.; Broermann, A.; Bretschneider, T.; Luippold, A.H.; Krawczyk, B.; Rist, W.; Simon, E. Biomarker discovery for chronic liver diseases by multi-omics—A preclinical case study. Sci. Rep. 2020, 10, 1314. [Google Scholar] [CrossRef]
- Messner, C.B.; Demichev, V.; Wendisch, D.; Michalick, L.; White, M.; Freiwald, A.; Textoris-Taube, K.; Vernardis, S.I.; Egger, A.S.; Kreidl, M.; et al. Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst. 2020, 11, 11–24.e14. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J. Systems biology elucidates common pathogenic mechanisms between nonalcoholic and alcoholic-fatty liver disease. PLoS ONE 2013, 8, e58895. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Nidhina Haridas, P.A.; Dwivedi, O.P.; Hyötyläinen, T.; Ali, A.; Juuti, A.; Leivonen, M.; Tukiainen, T.; Ahonen, L.; et al. Impaired hepatic lipid synthesis from polyunsaturated fatty acids in TM6SF2 E167K variant carriers with NAFLD. J. Hepatol. 2017, 67, 128–136. [Google Scholar] [CrossRef]
- Zhong, G.; Kirkwood, J.; Won, K.J.; Tjota, N.; Jeong, H.; Isoherranen, N. Characterization of Vitamin A Metabolome in Human Livers with and Without Nonalcoholic Fatty Liver Disease. J. Pharmacol. Exp. Ther. 2019, 370, 92–103. [Google Scholar] [CrossRef]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461.e1447. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Ajmera, V.H.; Puri, P.; Hsu, C.L.; Bassirian, S.; Mgdsyan, M.; Singh, S.; Faulkner, C.; Valasek, M.A.; Rizo, E.; et al. Serum metabolites detect the presence of advanced fibrosis in derivation and validation cohorts of patients with non-alcoholic fatty liver disease. Gut 2019, 68, 1884–1892. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Liu, L.; Guo, F.; Li, S.; Huang, L.; Sun, C.; Feng, R. Serum metabonomics of NAFLD plus T2DM based on liquid chromatography–mass spectrometry. Clin. Biochem. 2016, 49, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Mayo, R.; Crespo, J.; Martínez-Arranz, I.; Banales, J.M.; Arias, M.; Mincholé, I.; Aller de la Fuente, R.; Jimenez-Agüero, R.; Alonso, C.; de Luis, D.A.; et al. Metabolomic-based noninvasive serum test to diagnose nonalcoholic steatohepatitis: Results from discovery and validation cohorts. Hepatol. Commun. 2018, 2, 807–820. [Google Scholar] [CrossRef]
- Sansone, S.-A.; Fan, T.; Goodacre, R.; Griffin, J.L.; Hardy, N.W.; Kaddurah-Daouk, R.; Kristal, B.S.; Lindon, J.; Mendes, P.; Morrison, N.; et al. The Metabolomics Standards Initiative. Nat. Biotechnol. 2007, 25, 846–848. [Google Scholar] [CrossRef]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Perakakis, N.; Polyzos, S.A.; Yazdani, A.; Sala-Vila, A.; Kountouras, J.; Anastasilakis, A.D.; Mantzoros, C.S. Non-invasive diagnosis of non-alcoholic steatohepatitis and fibrosis with the use of omics and supervised learning: A proof of concept study. Metab. Clin. Exp. 2019, 101, 154005. [Google Scholar] [CrossRef]
- Khusial, R.D.; Cioffi, C.E.; Caltharp, S.A.; Krasinskas, A.M.; Alazraki, A.; Knight-Scott, J.; Cleeton, R.; Castillo-Leon, E.; Jones, D.P.; Pierpont, B.; et al. Development of a Plasma Screening Panel for Pediatric Nonalcoholic Fatty Liver Disease Using Metabolomics. Hepatol. Commun. 2019, 3, 1311–1321. [Google Scholar] [CrossRef]
- Luukkonen, P.K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1167–1175. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Scian, R.; Fernández Gianotti, T.; Dopazo, H.; Rohr, C.; Gaj, G.; San Martino, J.; Sevic, I.; Flichman, D.; et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. Am. J. Clin. Nutr. 2016, 103, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Banton, S.; Tran, V.T.; Konomi, J.V.; Li, S.; Jones, D.P.; Vos, M.B. Amino Acid Metabolism is Altered in Adolescents with Nonalcoholic Fatty Liver Disease-An Untargeted, High Resolution Metabolomics Study. J. Pediatrics 2016, 172, 14–19.e15. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Liu, X.; Zhou, K.; He, X.; Lu, C.; He, B.; Niu, X.; Xiao, C.; Xu, G.; Bian, Z.; et al. The Potential Biomarkers to Identify the Development of Steatosis in Hyperuricemia. PLoS ONE 2016, 11, e0149043. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schönauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects with Non-alcoholic Fatty Liver. Am. J. Gastroenterol 2017, 112, 102–110. [Google Scholar] [CrossRef]
- Zhou, Y.; Orešič, M.; Leivonen, M.; Gopalacharyulu, P.; Hyysalo, J.; Arola, J.; Verrijken, A.; Francque, S.; Van Gaal, L.; Hyötyläinen, T.; et al. Noninvasive Detection of Nonalcoholic Steatohepatitis Using Clinical Markers and Circulating Levels of Lipids and Metabolites. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016, 14, 1463–1472.e1466. [Google Scholar] [CrossRef] [PubMed]
- Chiappini, F.; Coilly, A.; Kadar, H.; Gual, P.; Tran, A.; Desterke, C.; Samuel, D.; Duclos-Vallée, J.C.; Touboul, D.; Bertrand-Michel, J.; et al. Metabolism dysregulation induces a specific lipid signature of nonalcoholic steatohepatitis in patients. Sci. Rep. 2017, 7, 46658. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Zhan, Z.Y.; Cao, H.Y.; Wu, C.; Bian, Y.Q.; Li, J.Y.; Cheng, G.H.; Liu, P.; Sun, M.Y. Urinary metabolomics analysis identifies key biomarkers of different stages of nonalcoholic fatty liver disease. World J. Gastroenterol. WJG 2017, 23, 2771–2784. [Google Scholar] [CrossRef] [PubMed]
- Troisi, J.; Pierri, L.; Landolfi, A.; Marciano, F.; Bisogno, A.; Belmonte, F.; Palladino, C.; Guercio Nuzio, S.; Campiglia, P.; Vajro, P. Urinary Metabolomics in Pediatric Obesity and NAFLD Identifies Metabolic Pathways/Metabolites Related to Dietary Habits and Gut-Liver Axis Perturbations. Nutrients 2017, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Caruso, M.G.; Tutino, V.; Bonfiglio, C.; Cozzolongo, R.; Giannuzzi, V.; De Nunzio, V.; De Leonardis, G.; Abbrescia, D.I.; Franco, I.; et al. Significant decrease of saturation index in erythrocytes membrane from subjects with non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 2017, 16, 160. [Google Scholar] [CrossRef]
- Qi, S.; Xu, D.; Li, Q.; Xie, N.; Xia, J.; Huo, Q.; Li, P.; Chen, Q.; Huang, S. Metabonomics screening of serum identifies pyroglutamate as a diagnostic biomarker for nonalcoholic steatohepatitis. Clin. Chim. Acta Int. J. Clin. Chem. 2017, 473, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, C.; Bullò, M.; Tinahones, F.J.; Martínez-González, M.; Corella, D.; Fragkiadakis, G.A.; López-Miranda, J.; Estruch, R.; Fitó, M.; Salas-Salvadó, J. Serum metabolites in non-alcoholic fatty-liver disease development or reversion; a targeted metabolomic approach within the PREDIMED trial. Nutr. Metab. 2017, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-X.; Hu, C.-X.; Sun, W.-L.; Pan, Q.; Shen, F.; Yang, Z.; Su, Q.; Xu, G.-W.; Fan, J.-G. Serum Monounsaturated Triacylglycerol Predicts Steatohepatitis in Patients with Non-alcoholic Fatty Liver Disease and Chronic Hepatitis B. Sci. Rep. 2017, 7, 10517. [Google Scholar] [CrossRef]
- Hu, X.-Y.; Li, Y.; Li, L.-Q.; Zheng, Y.; Lv, J.-H.; Huang, S.-C.; Zhang, W.; Liu, L.; Zhao, L.; Liu, Z.; et al. Risk factors and biomarkers of non-alcoholic fatty liver disease: An observational cross-sectional population survey. BMJ Open 2018, 8, e019974. [Google Scholar] [CrossRef]
- Tiwari-Heckler, S.; Gan-Schreier, H.; Stremmel, W.; Chamulitrat, W.; Pathil, A. Circulating Phospholipid Patterns in NAFLD Patients Associated with a Combination of Metabolic Risk Factors. Nutrients 2018, 10, 649. [Google Scholar] [CrossRef]
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986. [Google Scholar] [CrossRef] [PubMed]
- de Mello, V.D.; Sehgal, R.; Männistö, V.; Klåvus, A.; Nilsson, E.; Perfilyev, A.; Kaminska, D.; Miao, Z.; Pajukanta, P.; Ling, C.; et al. Serum aromatic and branched-chain amino acids associated with NASH demonstrate divergent associations with serum lipids. Liver Int. 2020, 41, 754–763. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Räsänen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietiläinen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e555. [Google Scholar] [CrossRef] [PubMed]
- Ægidius, H.M.; Veidal, S.S.; Feigh, M.; Hallenborg, P.; Puglia, M.; Pers, T.H.; Vrang, N.; Jelsing, J.; Kornum, B.R.; Blagoev, B.; et al. Multi-omics characterization of a diet-induced obese model of non-alcoholic steatohepatitis. Sci. Rep. 2020, 10, 1148. [Google Scholar] [CrossRef] [PubMed]
- Wruck, W.; Kashofer, K.; Rehman, S.; Daskalaki, A.; Berg, D.; Gralka, E.; Jozefczuk, J.; Drews, K.; Pandey, V.; Regenbrecht, C.; et al. Multi-omic profiles of human non-alcoholic fatty liver disease tissue highlight heterogenic phenotypes. Sci. Data 2015, 2, 150068. [Google Scholar] [CrossRef]
- Mesnage, R.; Biserni, M.; Balu, S.; Frainay, C.; Poupin, N.; Jourdan, F.; Wozniak, E.; Xenakis, T.; Mein, C.A.; Antoniou, M.N. Integrated transcriptomics and metabolomics reveal signatures of lipid metabolism dysregulation in HepaRG liver cells exposed to PCB 126. Arch. Toxicol 2018, 92, 2533–2547. [Google Scholar] [CrossRef]
- Mesnage, R.; Renney, G.; Séralini, G.E.; Ward, M.; Antoniou, M.N. Multiomics reveal non-alcoholic fatty liver disease in rats following chronic exposure to an ultra-low dose of Roundup herbicide. Sci. Rep. 2017, 7, 39328. [Google Scholar] [CrossRef]
- Qian, M.; Hu, H.; Yao, Y.; Zhao, D.; Wang, S.; Pan, C.; Duan, X.; Gao, Y.; Liu, J.; Zhang, Y.; et al. Coordinated changes of gut microbiome and lipidome differentiates nonalcoholic steatohepatitis (NASH) from isolated steatosis. Liver Int. 2020, 40, 622–637. [Google Scholar] [CrossRef]
- Jha, P.; McDevitt, M.T.; Gupta, R.; Quiros, P.M.; Williams, E.G.; Gariani, K.; Sleiman, M.B.; Diserens, L.; Jochem, A.; Ulbrich, A.; et al. Systems Analyses Reveal Physiological Roles and Genetic Regulators of Liver Lipid Species. Cell Syst. 2018, 6, 722–733.e726. [Google Scholar] [CrossRef]
- Lee, S.; Zhang, C.; Liu, Z.; Klevstig, M.; Mukhopadhyay, B.; Bergentall, M.; Cinar, R.; Ståhlman, M.; Sikanic, N.; Park, J.K.; et al. Network analyses identify liver-specific targets for treating liver diseases. Mol. Syst. Biol. 2017, 13, 938. [Google Scholar] [CrossRef]
- Chella Krishnan, K.; Kurt, Z.; Barrere-Cain, R.; Sabir, S.; Das, A.; Floyd, R.; Vergnes, L.; Zhao, Y.; Che, N.; Charugundla, S.; et al. Integration of Multi-omics Data from Mouse Diversity Panel Highlights Mitochondrial Dysfunction in Non-alcoholic Fatty Liver Disease. Cell Syst. 2018, 6, 103–115.e107. [Google Scholar] [CrossRef]
- Kurt, Z.; Barrere-Cain, R.; LaGuardia, J.; Mehrabian, M.; Pan, C.; Hui, S.T.; Norheim, F.; Zhou, Z.; Hasin, Y.; Lusis, A.J.; et al. Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol. Sex Differ. 2018, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.-Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e645. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Chappell, K.; Washam, C.L.; Gies, A.; Bird, J.; Robeson, M.S., 2nd; Byrum, S.D. Multi-omics data integration considerations and study design for biological systems and disease. Mol. Omics 2021, 17, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Artrith, N.; Butler, K.T.; Coudert, F.-X.; Han, S.; Isayev, O.; Jain, A.; Walsh, A. Best practices in machine learning for chemistry. Nat. Chem. 2021, 13, 505–508. [Google Scholar] [CrossRef]
- Alexandrov, T. Spatial Metabolomics and Imaging Mass Spectrometry in the Age of Artificial Intelligence. Annu. Rev. Biomed. Data Sci. 2020, 3, 61–87. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).