1. Introduction
Congenital anomalies of the kidneys and urinary tract (CAKUT) are the leading cause of chronic kidney disease (CKD) in children, and a large part of them is related to congenital obstructive nephropathy (CON) [
1,
2]. The pathogenesis of CON is complex and results from various functional and morphological changes associated with impaired urinary outflow. Obstruction in the urine flow causes an increase in the pressure in the urinary tract and leads to tubular dilatation. Renal response to mechanical compression involves angiotensin (Ang)-dependent renal vasoconstriction, ischemia, hypoxia, and accumulation of reactive oxygen species (ROS) [
3]. These are mediated by altered expression of growth factors, cytokines, including transforming growth factor-β1 (TGF-β1) and adhesion molecules [
3,
4,
5]. Functional lesions lead to cell apoptosis, interstitial macrophage recruitment, epithelial–mesenchymal transition (EMT), fibroblast–myofibroblast transformation, and accumulation of extracellular matrix (ECM) proteins. Morphological changes are responsible for interstitial fibrosis, tubular and vascular atrophy, and glomerular sclerosis. CON leads to the reduction of functioning nephrons. It causes glomerular hyperfiltration and consequent glomerular sclerosis of the remaining nephrons and is responsible for the occurrence of CKD in childhood or adulthood [
2,
6].
Widely used imaging studies such as ultrasonography (US) and scintigraphy and currently available laboratory indices of the kidney injury such as serum creatinine (Cr), cystatin C, estimated glomerular filtration rate (GFR), and urinary albumin excretion do not provide information about the early stage of renal fibrosis and are poor predictors of the future course of CON [
2,
7]. Therefore, unfavorable prognosis in children with CON mandates a search for non-invasive fibrotic markers, which would enable early diagnosis of the fibrosis process, assessment of the severity of fibrosis, and early intervention in children at risk of renal injury progression.
TGF-β1 is a 25 kDA well-known cytokine that plays a pivotal role in renal fibrosis. It is secreted by epithelial cells, macrophages, and fibroblasts as an inactive latent complex [
5,
8]. After activation, TGF-β1 binds to its type II receptor (TβRII), which recruits two types I receptors (TβRI)—Activin like kinase 1 (ALK1) and ALK5. Depending on the TβRI recruited, different kinds of Smad proteins become phosphorylated for cellular response to TGF-β1. The complex composed of TβRII and ALK1 leads to Smad1/5/8 phosphorylation, and the complex composed of TβRII and ALK5 leads to Smad2/3 phosphorylation. Smad cascade is one of the most essential and well-known pathways in progressive renal fibrosis [
9,
10,
11]. TGF-β1 promotes fibrosis by cell apoptosis, increasing macrophage infiltration, EMT, and myofibroblast transformation [
3]. Overexpression of TGF-β1 mRNA and protein was demonstrated in multiple models of renal fibrosis [
5,
12,
13]. Increased urinary TGF-β1 levels were found in CKD [
14] and various types of nephropathies, including glomerular (GN), diabetic (DN), and obstructive nephropathy [
8,
15,
16,
17].
Endoglin is a 180 kDa homodimeric protein consisting of extracellular, transmembrane, and intracellular domain. Endoglin expression was identified in endothelial, smooth muscle and mesangial cells, macrophages, and fibroblasts [
9,
18,
19]. Ang II and TGF-β1 are the most important stimulators of endoglin expression [
18,
20]. Endoglin acts as a TGF-β1 co-receptor known as TβRIII and binds to TGF-β1 by ALK1 and ALK5 receptors [
18]. It modulates cellular responses to TGF-β1, mainly related to myofibroblast transformation and ECM accumulation [
11,
20]. Cytoplasmatic domain differentiates two endoglin isoforms, named large (L-Eng) and short (S-Eng), according to the length of their cytoplasmatic tails. Upregulation of L-Eng leads to Smad1 and Smad2/3 phosphorylation and promotes fibrosis, while upregulation of S-Eng acts oppositely [
9,
19]. Overexpression of endoglin was shown in renal fibrosis models [
21,
22,
23] and in various human diseases, including CKD and DN [
11,
24].
Periostin is a 90 kDa matricellular protein with a multi-domain structure [
25,
26,
27]. It is induced during organogenesis and found only in small amounts in a healthy kidney. Periostin was expressed de novo during the chronic disease of several organs [
26,
28,
29]. In the kidney, periostin is released from fibroblasts and myofibroblasts in response to many factors, including Ang II and TGF-β1, which stimulate periostin expression by Smad phosphorylation and through Smad independent pathways [
25,
30]. It is expressed mainly in the location of tissue damage in ECM areas and directly interacts with multiple ECM proteins such as integrin, fibronectin, collagens, tenascin C, and bone morphogenic protein-1 (BMP-1) [
27,
28]. After binding to integrin, periostin induces fibrosis by increasing inflammatory cell infiltration, ECM remodeling, and TGF-β1 pathway upregulation [
29,
31]. Overexpression of periostin was reported in numerous experimental and clinical studies of renal fibrosis [
25,
32]. Increased urinary periostin levels were described in CKD, polycystic kidney disease (PKD), and various types of nephropathies, including IgA nephropathy (IgAN) [
29,
32,
33].
Cytokeratin-18 (CK-18) is a 45 kDA cytoskeletal protein consisting of three domains [
34,
35]. It is a family member of intermediate filament proteins and represents about 5% of total cell proteins in most epithelial and parenchymal cells [
36,
37]. CK-18 is involved in maintaining cell shape and integrity, mechanical stability, intracellular organization, and cell signaling, transport, and differentiation [
35,
38,
39]. In addition, it protects cells from mechanical and non-mechanical stress [
35,
40]. In response to various stress situations, CK-18 undergoes phosphorylation that modifies its solubility, filaments organization, prevents interaction with other molecules, and protects from proteolysis and degradation [
35,
39]. Overexpression of CK-18 was found in models of renal fibrosis and in humans with diabetic and lupus nephropathy [
39]. High urinary CK-18 levels were reported in acute kidney injury (AKI) and CKD [
37,
39].
To the best of our knowledge, there are no studies in the available literature investigating the usefulness of urinary endoglin, periostin, and CK-18 in children with CON. Therefore, the aim of our study was to investigate the predictive value of urinary endoglin, periostin, CK-18, and TGF-β1 for assessing the severity of renal fibrosis in children with congenital obstructive nephropathy.
4. Discussion
Our cross-sectional single-center study investigated the predictive value of new biomarkers, such as endoglin, periostin, and CK-18, for assessing the severity of renal scarring in children with CON. We also evaluated TGF-β1, the most recognized fibrotic factor, which served as our reference (“gold standard”). Here, we report that urinary periostin, periostin/Cr, and CK-18 were significantly higher in children with CON compared to the healthy controls. In addition, children with severe scars had significantly higher urinary periostin/Cr levels than those with borderline lesions. Thus, periostin and periostin/Cr demonstrated a better diagnostic profile for diagnosing renal scars than other evaluated biomarkers. However, both periostin and periostin/Cr did not differentiate severe and moderate scars from borderline lesions. Endoglin showed moderate usefulness for diagnosing borderline lesions, and CK-18 and TGF-β1 revealed low utility for diagnosing severe and moderate scars.
Multiple clinical studies confirmed the usefulness of urinary TGF-β1 for the diagnosis of urinary tract obstruction and tubulointerstitial fibrosis. Children with obstructive uropathy demonstrated significantly higher TGF-β1 levels than those with non-obstructive hydronephrosis and healthy controls [
4,
45,
46]. A significant decrease in elevated TGF-β1 levels was observed after pyeloplasty [
47]. Patients who improved renal function after nephrostomy (i.e., those in whom GFR increased) had significantly lower TGF-β1 levels at presentation compared to those who did not [
48]. Elevated urinary TGF-β1 demonstrated 82% accuracy for differentiation reversible obstruction from irreversible [
48] and 90.8% accuracy for long-term follow-up after pyeloplasty [
47]. The ESCAPE trial revealed markedly higher urinary TGF-β1 levels in children with mild to moderate CKD and CON compared to those with CKD and other kidney diseases [
14].
In contrast, our study found no differences in urinary TGF-β1 and TGF-β1/Cr levels between children with CON and healthy controls. Palmer et al. reported elevated TGF-β1 levels only in the pelvis of the obstructed kidney but not in the bladder urine [
49]. Some authors suppose that the production of TGF-β1 is increased in the early phase of tissue damage and reduced in the terminal stage of tissue degeneration [
14,
17]. We did not find any differences in TGF-β1 and TGF-β1/Cr levels between subgroups with severe, moderate scars and borderline lesions. Despite this, urinary TGF-β1 was associated with the presence of advanced scars. However, its diagnostic potential for detecting scars was low. In our study, 17% of children were at least one year after surgery of urinary tract obstruction, which could reduce their TGF-β1 level. A slight increase in serum Cr was found in 7.4% of children and in cystatin C in 18.5% of children. RRF in scintigraphy less than 10% demonstrated in 2.5% of patients. It could also have had an impact on the results obtained.
The importance of endoglin in renal fibrosis was documented in several animal models of renal fibrosis. For example, the model of tubulointerstitial fibrosis induced by unilateral ureteral obstruction (UUO) demonstrated an increase in endoglin mRNA and protein expression in the obstructed kidney [
9,
19,
50]. Model of renal ischemia–reperfusion (I-R) injury showed coincidence endoglin expression with increased TGF-β1 mRNA expression. In this study, haploinsufficient mice of endoglin (Eng+/−) were protected for renal I-R injury compared to their wild type (WT) littermates (Eng+/+) [
20]. Roy-Chaudhury et al. described an association of interstitial endoglin expression and chronic histological damage in biopsies from patients with progressive CKD [
24]. A recent study by Gerrits et al. revealed an increase in interstitial endoglin expression in autopsy samples obtained from diabetic patients with DN compared to those without DN. Renal endoglin expression correlated with the degree of interstitial fibrosis and increased serum Cr, reduced GFR, and hypertension in DN [
11].
In our cohort of pediatric patients, we demonstrated no differences in urinary endoglin and endoglin/Cr levels between children with CON and the controls, as well as between subgroups with severe, moderate scars, and borderline lesions. Only endoglin/Cr was related to the presence of borderline lesions. In ROC analysis, endoglin showed moderate utility as a biomarker for diagnosing borderline lesions. However, its specificity was very high. We did not evaluate L-Eng and S-Eng isoforms. Therefore, we do not know if an association of endoglin with borderline lesions is due to the upregulation of pro-fibrotic L-Eng or anti-fibrotic S-Eng, especially in children with borderline lesions.
Recent studies have identified periostin as a novel key factor in the progression of kidney disease. Overexpression of periostin following renal injury was described in different models of renal fibrosis, such as UUO, 5/6 nephrectomy, hypertensive renal injury, and renal I-R injury [
31,
51,
52]. High expression of periostin was associated with acceleration of cyst growth and fibrosis in PKD [
33,
53]. Periostin overexpression was observed in patients with different types of progressive nephropathies, including lupus nephropathy and chronic allograft nephropathy (CAN) [
7,
32,
54]. Patients with high levels of urinary periostin at the time of AKI episode were more likely to progress to CKD [
52]. In the study by Hwang et al., higher urinary periostin/Cr levels at the time of renal biopsy were associated with tissue periostin overexpression, a higher degree of fibrosis, a greater decline in GFR, and poor renal outcome in IgAN patients [
29]. Some studies demonstrated a protective effect of periostin suppression for CKD progression. Periostin-null mice showed a decrease in apoptosis compared to the WT mice [
25,
52].
In the present study, we demonstrated significantly higher urinary periostin and periostin/Cr levels in children with CON compared to the controls and significantly higher periostin/Cr levels in children with severe scars compared to those with borderline lesions. Both periostin and periostin/Cr demonstrated relatively high utility for diagnosing scars and moderate utility for diagnosing borderline lesions. However, based on diagnostic profile and cut-off in ROC analysis, periostin did not allow for differentiation of severe and moderate scars from borderline lesions.
It is difficult to explain why periostin was of no use to differentiate scars from borderline lesions, especially that we showed significantly higher levels of periostin/Cr in children with severe scars compared to those with borderline lesions. Although we confirmed that periostin is strongly related to renal fibrosis, we still know too little about the regulation of periostin expression and its signaling pathways [
33]. Recent evidence suggests that periostin might be a tissue repair molecule that stimulates signaling pathways involved in tissue regeneration [
33,
55,
56]. Korman et al. revealed a protective effect of periostin in the model of AKI induced by renal I-R injury. They demonstrated that periostin overexpression was associated with lower expression of pro-inflammatory cytokines, less epithelial damage, increased proliferation of pro-regenerative macrophage phenotypes, and less severe injury than the periostin-null mouse. In the authors′ opinion, periostin may play a protective role in AKI and a detrimental role in CKD [
56]. In our study, we followed patients with chronic renal fibrosis. Therefore, periostin overexpression could not be related to renal tissue regeneration.
CK-18 is a cell-protective and stress-responsive protein that seems to be a novel sensitive marker of renal tubular cell stress and tubular injury. Experimental studies demonstrated an increase in CK-18 expression in different models of renal injury. In the model of UUO, upregulation of CK-18 occurred very early after UUO induction and increased with renal injury progression. Additionally, the model of progressive GN with secondary tubulointerstitial injury and fibrosis showed only slight CK-18 expression in the early stage of disease and significantly higher expression in the late stage. Overexpression of CK-18 in renal biopsy was reported in the majority of renal tubules in the cast, diabetic, and lupus nephropathy, as well in the Bowman capsule in crescentic necrotic GN [
39].
In this study, we found significantly higher urinary CK-18 levels in children with CON compared to the controls, but no differences were observed in terms of CK-18 and CK-18/Cr between the three subgroups. Urinary CK-18 was associated with the presence of scars, but its diagnostic value was low. In the study by Djudjaj et al., elevated urinary CK-18 levels were found in animals with adenine nephropathy and Alport syndrome and in patients with AKI [
39]. Roth et al. reported significantly higher urinary CK-18 levels in CKD stage 5 compared to the healthy controls [
37]. During cell degeneration, CK-18 is released in two forms depending on the type of cell death. In cells apoptosis, caspase cleaved CK-18 fragments are produced, while in cell necrosis, CK-18 is liberated without caspase-mediated modifications [
57]. In CKD, cell loss results mainly from cell necrosis, while in the obstructed kidney, cell death is most susceptible to apoptosis [
3,
58]. Choi et al. confirmed a progressive increase in tubular and interstitial cell apoptosis during the duration of renal obstruction [
59]. We did not assess caspase cleaved CK-18. However, we can speculate that our patients could have high levels of CK-18 fragments due to apoptosis followed by urinary tract obstruction.
It is not known whether urinary fibrotic biomarkers are associated with clinical and laboratory indices of kidney injury. In the ESCAPE trial, GFR inversely correlated with urinary TGF-β1 [
14]. Similarly, a negative correlation was reported with urinary periostin in IgAN [
29] and chronic allograft nephropathy (CAN) [
7] and urinary CK-18 in CKD [
37]. Some studies reported positive correlations of TGF-β1 with proteinuria and urinary α1-microglobulin in CON [
4], urinary periostin with proteinuria in CAN [
7], and CK-18 with proteinuria and albuminuria in CKD stage 5 [
37]. In contrast, other authors did not show any correlations of urinary TGF-β1 with serum Cr, GFR, ACR, proteinuria, and degree of tubulointerstitial fibrosis [
4,
17].
We found positive correlations of the normalized biomarkers with cystatin C and ACR, negative correlations with serum Cr, and no relation with GFR except endoglin/Cr. These negative correlations with serum Cr were probably related to our study′s relatively large number of young children. In infants and young children, low serum Cr levels, due to small muscle mass in this age, and greater Cr tubular reabsorption lead to low urinary Cr excretion [
2]. It results in higher values of normalized biomarkers and is responsible for a negative correlation with serum Cr. Due to the heterogenicity of children′s age in our study, we assessed biomarkers with and without normalization.
In our study, we also evaluated the association of widely used laboratory indices of kidney injury with the presence of renal scars and borderline lesions. We found that only ACR was able to differentiate children with scars from those with borderline lesions. However, its diagnostic value was relatively low. The ESCAPE trial also demonstrated inter-correlation of urinary TGF-β1 with other biomarkers that participated in the progression of tissue injury in CKD [
14]. In line with this study, we found an inter-correlation of all normalized biomarkers. These results are not unexpected. It is well-known that TGF-β1 is the major stimulator of endoglin and periostin expression, periostin creates a feedback loop with TGF-β1, and CK18 was identified as an important factor in renal fibrosis [
9,
10,
27,
32,
33,
39].
Some studies evaluated the correlation of the intensity of renal fibrosis and urinary biomarkers with RRF of obstructed kidney in a nuclear renal scan. Zhang demonstrated a correlation between the intensity of renal fibrosis in renal biopsy performed during pyeloplasty and RRF of affected kidney [
60]. In contrast, other authors did not show any correlation between urinary TGF-β1 and RRF [
4,
47]. In the present study, 99mTc-EC scintigraphy was used for renal scars assessment, which is an alternative to 99mTc-dimercaptosuccinic acid (DMSA) scintigraphy, with high sensitivity (98.75%) and specificity (99.15%) for detection of scars in normally positioned kidneys but with lower radiation dose [
61]. We demonstrated that approximately half of our children with advanced scars demonstrated tracer uptake higher than 45%. We also did not find a correlation between urinary biomarkers and RRF in children with CON.
Urinary tract obstruction leads to a reduction in functioning nephrons and to the compensatory adaptation of the remaining. Therefore, it may affect RRF results [
2]. In addition, in hydronephrosis, tracer accumulation in the obstructed kidney, as well as an increase in blood flow caused by altered renal hemodynamics, may cause falsely high RRF [
62,
63]. Men-Meir et al. demonstrated that “supranormal” RRF in the obstructed kidney is not always a favorable sign and may even be a warning sign of impending decompensation. Therefore, a measure of RRT has a low predictive value for renal fibrosis in children with CON.
Our study has some limitations. This single-center research may be associated with the bias occurrence. The cross-sectional design of the study prevents from drawing definitive and causative conclusions. The number of analyzed patients in subgroups was relatively low. Heterogenicity of patients′ ages, various causes of obstructive nephropathy, and different patient follow-up may impact the results. In addition, we have not analyzed the fibrosis markers in other groups of patients, e.g., in glomerulopathies.


 ,
, 

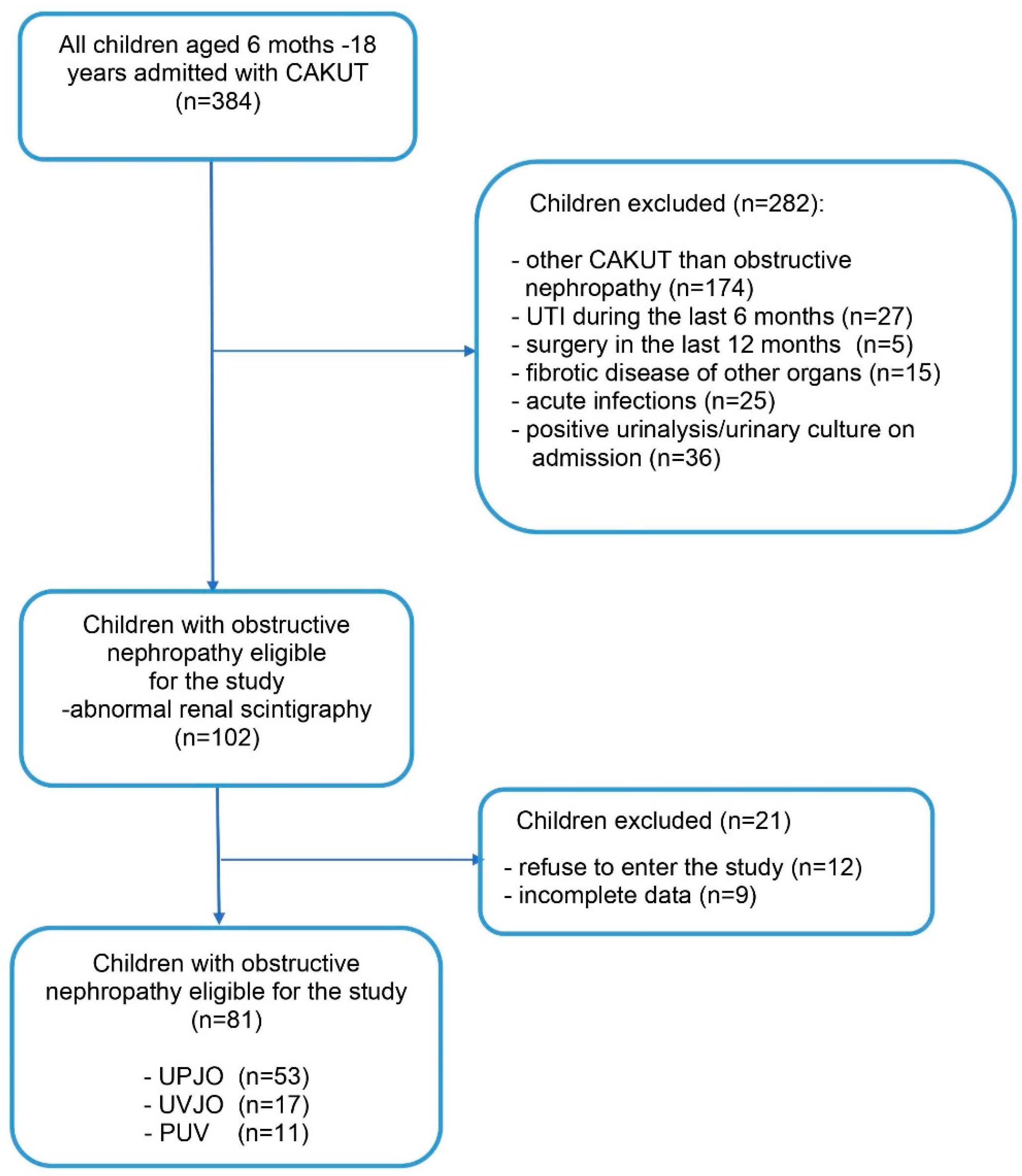{kind=link}