
DNA Methylation Changes in Fibromyalgia Suggest the Role of the Immune-Inflammatory Response and Central Sensitization

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Demographic and Clinical Assessment
2.3. Samples Collection
2.4. DNA Methylation Analysis
2.5. Bioinformatic Data Processing
2.6. Statistical Analyses
3. Results
3.1. Characteristics of the Study Participants
3.2. DNA Methylation Analysis Comparing Cases and Controls
3.3. Network Analysis
3.4. Cluster Analysis of the Socio-Demographic Variables
3.5. The Concurrent Effect of DNA Methylation, Depression, and the Clustered Sociodemographic Data on FM Risk
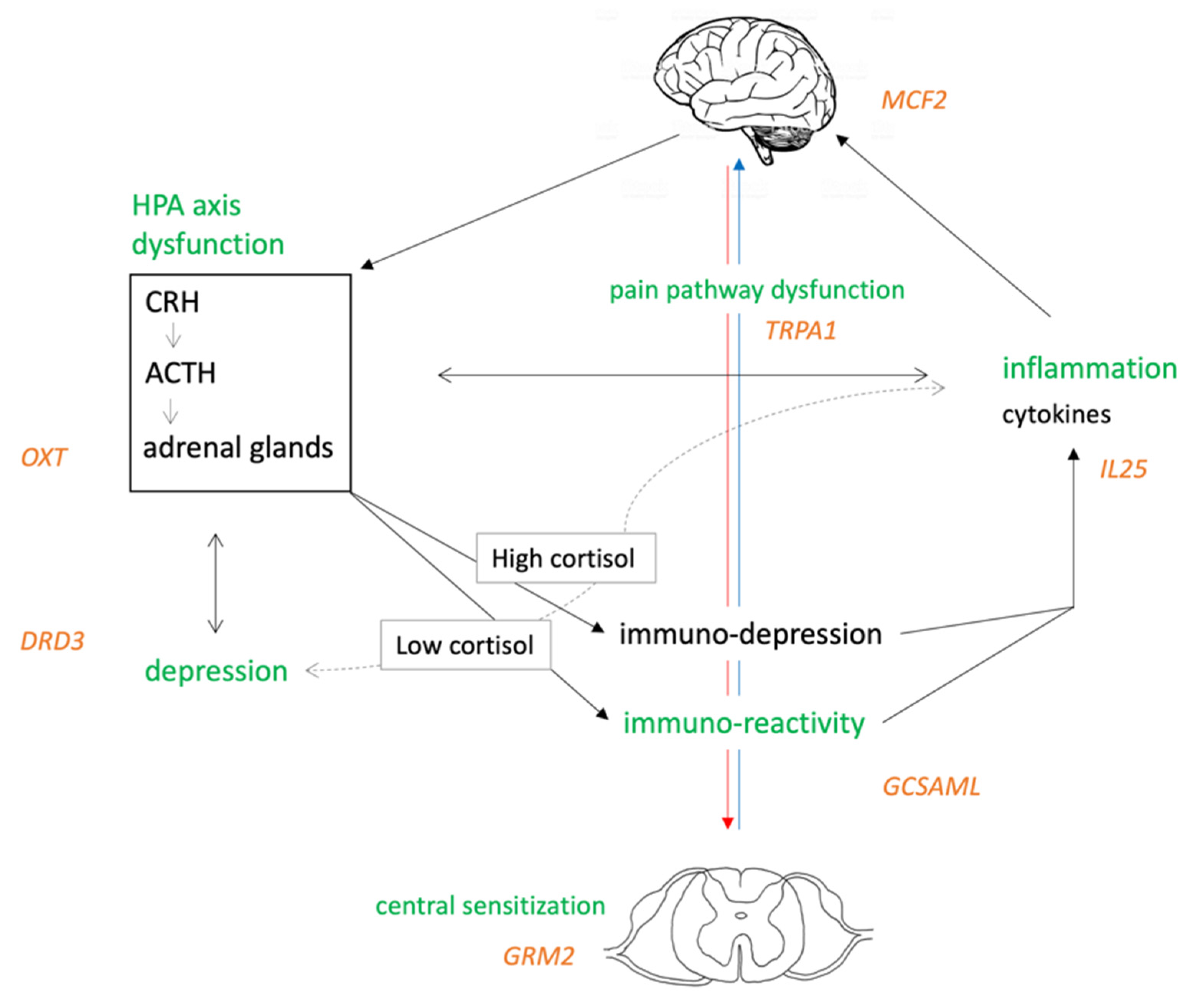
4. Discussion
4.1. DNA Methylation Analysis
4.2. Concomitant Risk Factors on FM Risk
4.3. Limitations and Future Research Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Katz, R.S.; Mease, P.; Russell, A.S.; Russell, I.J.; Winfield, J.B.; Yunus, M.B. The American College of Rheumatology preliminary diagnostic criteria for fibromyalgia and measurement of symptom severity. Arthritis Care Res. 2010, 62, 600–610. [Google Scholar] [CrossRef] [Green Version]
- Wolfe, F.; Clauw, D.J.; Fitzcharles, M.A.; Goldenberg, D.L.; Häuser, W.; Katz, R.L.; Mease, P.J.; Russell, A.S.; Russell, I.J.; Walitt, B. 2016 Revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin. Arthritis Rheum. 2016, 46, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Treede, R.-D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. Chronic pain as a symptom or a disease: The IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 2019, 160, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, L.A.; Henriksson, K.G. Pathophysiological mechanisms in chronic musculoskeletal pain (fibromyalgia): The role of central and peripheral sensitization and pain disinhibition. Best Pract. Res. Clin. Rheumatol. 2007, 21, 465–480. [Google Scholar] [CrossRef]
- Kosek, E.; Cohen, M.; Baron, R.; Gebhart, G.F.; Mico, J.-A.; Rice, A.S.C.; Rief, W.; Sluka, A.K. Do we need a third mechanistic descriptor for chronic pain states? Pain 2016, 157, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Coskun Benlidayi, I. Role of inflammation in the pathogenesis and treatment of fibromyalgia. Rheumatol. Int. 2019, 39, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Russell, I.J.; Stewart, J.M.; Gleason, R.M.; Theoharides, T.C. Neuropeptides CRH, SP, HK-1, and Inflammatory Cytokines IL-6 and TNF Are Increased in Serum of Patients with Fibromyalgia Syndrome, Implicating Mast Cells. J. Pharmacol. Exp. Ther. 2016, 356, 664–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofford, L.J.; Pillemer, S.R.; Kalogeras, K.T.; Cash, J.M.; Michelson, D.; Kling, M.A.; Sternberg, E.M.; Gold, P.W.; Chrousos, G.P.; Wilder, R.L. Hypothalamic–pituitary–adrenal axis perturbations in patients with fibromyalgia. Arthritis Rheum. 1994, 37, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, D.S.; MacKie, P.J.; Kareken, D.A.; Hutchins, G.D.; Chumin, E.J.; Christian, B.T.; Yoder, K.K. Differential dopamine function in fibromyalgia. Brain Imaging Behav. 2016, 10, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Ablin, J.N.; Buskila, D. Update on the genetics of the fibromyalgia syndrome. Best Pract. Res. Clin. Rheumatol. 2015, 29, 20–28. [Google Scholar] [CrossRef]
- D’Agnelli, S.; Arendt-Nielsen, L.; Gerra, M.C.; Zatorri, K.; Boggiani, L.; Baciarello, M.; Bignami, E. Fibromyalgia: Genetics and epigenetics insights may provide the basis for the development of diagnostic biomarkers. Mol. Pain 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Yavne, Y.; Amital, D.; Watad, A.; Tiosano, S.; Amital, H. A systematic review of precipitating physical and psychological traumatic events in the development of fibromyalgia. Semin. Arthritis Rheum. 2018, 48, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Deans, C.; Maggert, K.A. What do you mean, “epigenetic”? Genetics 2015, 199, 887–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.C.; Resendiz, M.; Lo, C.L. Environmental Influence of Epigenetics, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780128053881. [Google Scholar]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Gerra, M.C.; Carnevali, D.; Pedersen, I.S.; Donnini, C.; Manfredini, M.; González-Villar, A.; Triñanes, Y.; Pidal-Miranda, M.; Arendt-Nielsen, L.; Carrillo-de-la-Peña, M.T. DNA methylation changes in genes involved in inflammation and depression in fibromyalgia: A pilot study. Scand. J. Pain 2021, 21, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, C.; Clark, S.; Bennett, R. The fibromyalgia impact questionnaire: Development and validation. J. Rheumatol. 1991, 18, 728–733. [Google Scholar]
- Esteve-Vives, J.; Rivera Redondo, J.; Salvat Salvat, M.I.; de Gracia Blanco, M.; Alegre de Miguel, C. Propuesta de una versión de consenso del Fibromyalgia Impact Questionnaire (FIQ) para la población española. Reumatol. Clin. 2007, 3, 21–24. [Google Scholar] [CrossRef]
- McCormack, H.; Horne, D.; Sheather, S. Clinical applications of visual analogue scales: A critical review. Psychol. Med. 1988, 18, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Ward, C.; Mendelson, M.; Mock, J.; Erbaugh, J. An inventory for measuring depression. Arch. Gen. Psychiatry 1961, 4, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, J.; Vázquez, C. Fiabilidad, validez y datos normativos del Inventario para la Depresión de Beck. Psicothema 1998, 10, 303–318. [Google Scholar]
- Buysse, D.; Reynolds, C.; Monk, T.; Berman, S.; Kupfer, D. The Pittsburgh Sleep Quality Index: A New Instrument for Psychiatric Practice and Research. Phychiatry Res. 1988, 28, 193–213. [Google Scholar] [CrossRef]
- Macías, J.; Royuela, A. La versión española del Índice de Calidad de Sueño de Pittsburgh. Inf. Psiquiátricas 1996, 146, 465–472. [Google Scholar]
- Alvarado, S.; Tajerian, M.; Suderman, M.; Machnes, Z.; Pierfelice, S.; Millecamps, M.; Stone, L.S.; Szyf, M. An epigenetic hypothesis for the genomic memory of pain. Front. Cell. Neurosci. 2015, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Livshits, G.; Malkin, I.; Freidin, M.B.; Xia, Y.; Gao, F.; Wang, J.; Spector, T.D.; Macgregor, A.; Bell, J.T.; Williams, F.M.K. Genome-wide methylation analysis of a large population sample shows neurological pathways involvement in chronic widespread musculoskeletal pain. Pain 2017, 158, 1053–1062. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.T.; Loomis, A.K.; Butcher, L.M.; Gao, F.; Zhang, B.; Hyde, C.L.; Sun, J.; Wu, H.; Ward, K.; Harris, J.; et al. Differential methylation of the TRPA1 promoter in pain sensitivity. Nat. Commun. 2014, 5, 2978. [Google Scholar] [CrossRef] [Green Version]
- Sukenaga, N.; Ikeda-Miyagawa, Y.; Tanada, D.; Tunetoh, T.; Nakano, S.; Inui, T.; Satoh, K.; Okutani, H.; Noguchi, K.; Hirose, M. Correlation Between DNA Methylation of TRPA1 and Chronic Pain States in Human Whole Blood Cells. Pain Med. 2016, 17, 1906–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sá Machado Araújo, G.; da Silva Francisco Junior, R.; dos Santos Ferreira, C.; Mozer Rodrigues, P.T.; Terra Machado, D.; Louvain de Souza, T.; Teixeira de Souza, J.; Figueiredo Osorio da Silva, C.F.O.; Alves da Silva, A.F.; Andrade, C.C.F.; et al. Maternal 5 m CpG imprints at the PARD6G-AS1 and GCSAML differentially methylated regions are decoupled from parent-of-origin expression effects in multiple human tissues. Front. Genet. 2018, 9, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Z.; Zhu, L.-J.; Li, Y.-Q.; Hao, L.-Y.; Yin, C.; Yang, J.-X.; Guo, Y.; Zhang, S.; Hua, L.; Xue, Z.-Y.; et al. Epigenetic modification of spinal miR-219 expression regulates chronic inflammation pain by targeting CaMKIIgamma. J. Neurosci. 2014, 34, 9476–9483. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.D.; Gelbart, T.; Whisenant, T.C.; Waalen, J.; Mondala, T.S.; Ikle, D.N.; Salomon, D.R.; Bennett, R.M.; Kurian, S.M. Genome-wide expression profiling in the peripheral blood of patients with fibromyalgia. Clin. Exp. Rheumatol. 2016, 34, S89–S98. [Google Scholar] [PubMed]
- Teschler, S.; Bartkuhn, M.; Künzel, N.; Schmidt, C.; Kiehl, S.; Dammann, G.; Dammann, R. Aberrant methylation of gene associated CpG sites occurs in borderline personality disorder. PLoS ONE 2013, 8, e84180. [Google Scholar] [CrossRef]
- Notartomaso, S.; Mascio, G.; Bernabucci, M.; Zappulla, C.; Scarselli, P.; Cannella, M.; Imbriglio, T.; Gradini, R.; Battaglia, G.; Bruno, V.; et al. Analgesia induced by the epigenetic drug, L-acetylcarnitine, outlasts the end of treatment in mouse models of chronic inflammatory and neuropathic pain. Mol. Pain 2017, 13, 1744806917697009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, D.-Y.; Bai, G.; Ji, Y.; Traub, R.J. Epigenetic upregulation of metabotropic glutamate receptor 2 in the spinal cord attenuates oestrogen-induced visceral hypersensitivity. Gut 2015, 64, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Potvin, S.; Larouche, A.; Normand, E.; de Souza, J.B.; Gaumond, I.; Grignon, S.; Marchand, S. DRD3 Ser9Gly polymorphism is related to thermal pain perception and modulation in chronic widespread pain patients and healthy controls. J. Pain 2009, 10, 969–975. [Google Scholar] [CrossRef]
- Hong, S.; Zheng, G.; Wiley, J.W. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology 2015, 148, 148–157.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, R.; Dymov, S.; Millecamps, M.; Suderman, M.; Gregoire, S.; Koenigs, K.; Alvarado, S.; Tajerian, M.; Stone, L.S.; Szyf, M. Overlapping signatures of chronic pain in the DNA methylation landscape of prefrontal cortex and peripheral T cells. Sci. Rep. 2016, 6, 19615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.W.; Filkowski, M.M.; Cochran, R.N.; Denison, L.; Ishak, A.; Nishitani, S.; Smith, A.K. Epigenetic modification of OXT and human sociability. Proc. Natl. Acad. Sci. USA 2016, 113, E3816–E3823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.; Pellegrini, M. BS-Seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genom. 2013, 14, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Zhu, P.; Pellegrini, M.; Zhang, M.Q.; Wang, X.; Ni, Z. CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 2018, 34, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. Metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, G.; Franceschini, A. A multidimensional version of the Kolmogorov–Smirnov test. Mon. Not. R. Astron. Soc. 1987, 225, 155–170. [Google Scholar] [CrossRef]
- Takahashi, A.; Flanigan, M.E.; McEwen, B.S.; Russo, S.J. Aggression, Social Stress, and the Immune System in Humans and Animal Models. Front. Behav. Neurosci. 2018, 12, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meester, I.; Rivera-Silva, G.F.; González-Salazar, F. Immune System Sex Differences May Bridge the Gap Between Sex and Gender in Fibromyalgia. Front. Neurosci. 2019, 13, 1414. [Google Scholar] [CrossRef] [PubMed]
- de Vega, W.C.; Vernon, S.D.; McGowan, P.O. DNA methylation modifications associated with chronic fatigue syndrome. PLoS ONE 2014, 9, e104757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, R.; Ren, J.; Lewith, G.; Barclay, W.; Holgate, S.; Almond, J. Identification of novel expressed sequences, up-regulated in the leucocytes of chronic fatigue syndrome patients. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2003, 33, 1450–1456. [Google Scholar] [CrossRef]
- Vernon, S.D.; Unger, E.R.; Dimulescu, I.M.; Rajeevan, M.; Reeves, W.C. Utility of the blood for gene expression profiling and biomarker discovery in chronic fatigue syndrome. Dis. Markers 2002, 18, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovejero, T.; Sadones, O.; Sánchez-Fito, T.; Almenar-Pérez, E.; Espejo, J.A.; Martín-Martínez, E.; Nathanson, L.; Oltra, E. Activation of Transposable Elements in Immune Cells of Fibromyalgia Patients. Int. J. Mol. Sci. 2020, 21, 1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, M.S.; Oltra, E.; Sarria, L.; Rose, N.; Beljanski, V.; Fletcher, M.A.; Klimas, N.G.; Nathanson, L. Identification of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome-associated DNA methylation patterns. PLoS ONE 2018, 13, e0201066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagelberg, N.; Martikainen, I.K.; Mansikka, H.; Hinkka, S.; Någren, K.; Hietala, J.; Scheinin, H.; Pertovaara, A. Dopamine D2 receptor binding in the human brain is associated with the response to painful stimulation and pain modulatory capacity. Pain 2002, 99, 273–279. [Google Scholar] [CrossRef]
- Hirsch, E.; Pozzato, M.; Vercelli, A.; Barberis, L.; Azzolino, O.; Russo, C.; Vanni, C.; Silengo, L.; Eva, A.; Altruda, F. Defective dendrite elongation but normal fertility in mice lacking the Rho-like GTPase activator Dbl. Mol. Cell. Biol. 2002, 22, 3140–3148. [Google Scholar] [CrossRef] [Green Version]
- Docampo, E.; Escaramís, G.; Gratacòs, M.; Villatoro, S.; Puig, A.; Kogevinas, M.; Collado, A.; Carbonell, J.; Rivera, J.; Vidal, J.; et al. Genome-wide analysis of single nucleotide polymorphisms and copy number variants in fibromyalgia suggest a role for the central nervous system. Pain 2014, 155, 1102–1109. [Google Scholar] [CrossRef]
- Heinrichs, M.; Baumgartner, T.; Kirschbaum, C.; Ehlert, U. Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol. Psychiatry 2003, 54, 1389–1398. [Google Scholar] [CrossRef]
- Uvnäs-Moberg, K.; Bruzelius, G.; Alster, P.; Lundeberg, T. The antinociceptive effect of non-noxious sensory stimulation is mediated partly through oxytocinergic mechanisms. Acta Physiol. Scand. 1993, 149, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Arletti, R.; Benelli, A.; Bertolini, A. Influence of oxytocin on nociception and morphine antinociception. Neuropeptides 1993, 24, 125–129. [Google Scholar] [CrossRef]
- Durand, D.; Pampillo, M.; Caruso, C.; Lasaga, M. Role of metabotropic glutamate receptors in the control of neuroendocrine function. Neuropharmacology 2008, 55, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Eller-Smith, O.C.; Nicol, A.L.; Christianson, J.A. Potential Mechanisms Underlying Centralized Pain and Emerging Therapeutic Interventions. Front. Cell. Neurosci. 2018, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, R.; Mork, P.J.; Westgaard, R.H.; Rø, M.; Lundberg, U. Fibromyalgia syndrome is associated with hypocortisolism. Int. J. Behav. Med. 2010, 17, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Thomson, C.A.; McColl, A.; Cavanagh, J.; Graham, G.J. Peripheral inflammation is associated with remote global gene expression changes in the brain. J. NeuroInflamm. 2014, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar]
- Keller, J.; Gomez, R.; Williams, G.; Lembke, A.; Lazzeroni, L.; Murphy, G.M.J.; Schatzberg, A.F. HPA axis in major depression: Cortisol, clinical symptomatology and genetic variation predict cognition. Mol. Psychiatry 2017, 22, 527–536. [Google Scholar] [CrossRef] [PubMed]
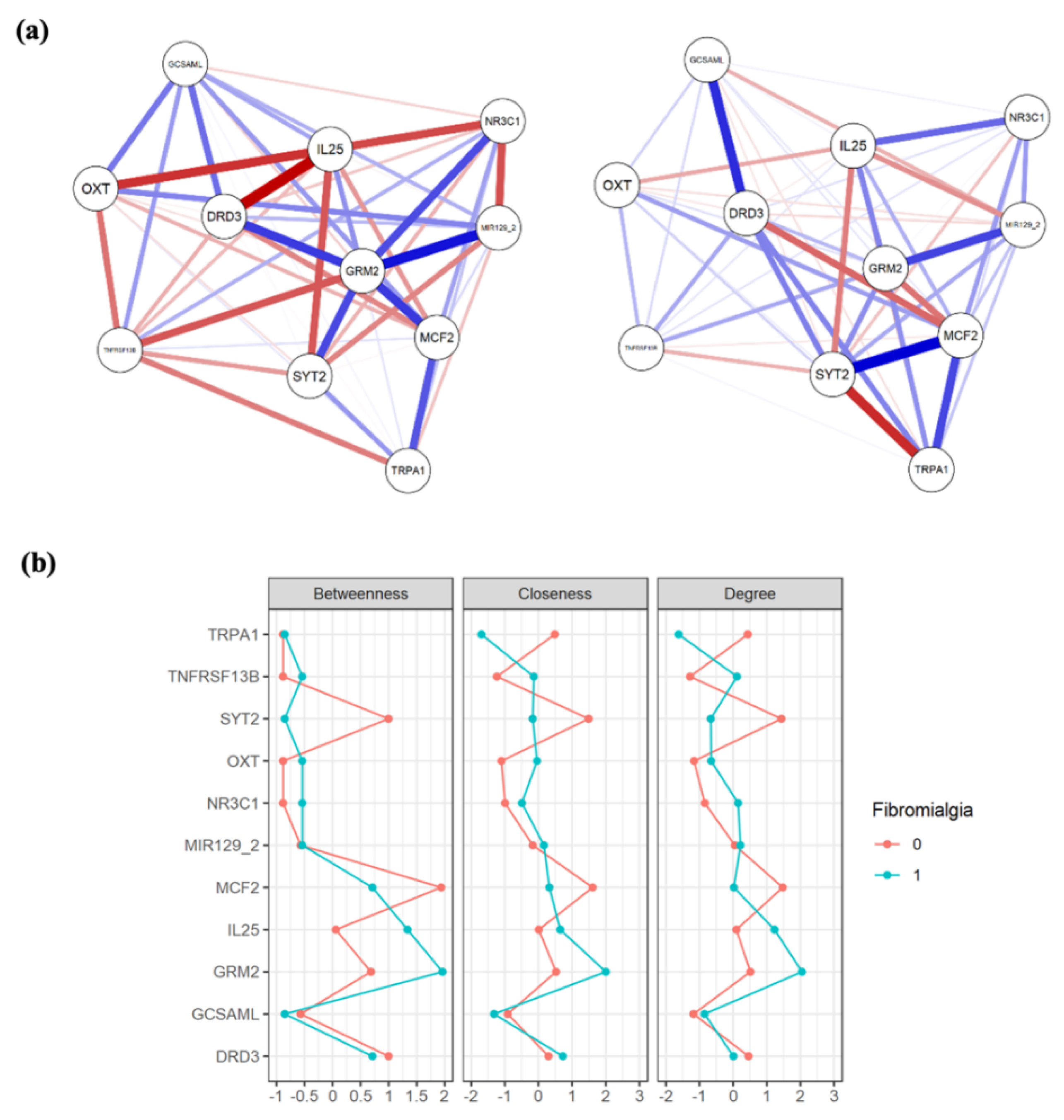

{kind=link}
{kind=link}
| Gene | Chr | Target Region | Relevant Association | PMID | |
|---|---|---|---|---|---|
| Start | End | ||||
| SYT2 | chr1 | 202709820 | 202709921 | Long-term changes in DNA methylation in the PFC in a chronic pain model. | 25852480 [24] |
| TNFRSF13B | chr17 | 16971786 | 16972349 | DNA methylation changes associated to chronic widespread musculoskeletal pain. | 28221285 [25] |
| TRPA1 | chr8 | 72075180 | 72076496 | Differentially methylated regions associated with high or low pain sensitivity and with chronic pain. | 24496475 [26] 26849948 [27] |
| GCSAML | chr1 | 247518380 | 247518621 | In a DMR, maternally inherited 5mCpG imprints with potential influence on transcription factors expression from the paternal allele. | 29545821 [28] |
| MIR129-2 | chr11 | 43581119 | 43581338 | Hypermethylation of CpG islands in the miR-219 promoter in a chronic inflammation pain model. | 25031391 [29] |
| IL25 | chr14 | 23372249 | 23372369 | Differentially expressed comparing FM patients and controls. | 27157394 [30] |
| MCF2 | chr X | 139692217 | 139692357 | DNA methylation changes in borderline personality disorder. | 24367640 [31] |
| GRM2 | chr3 | 51706813 | 51707270 | Epigenetically regulation of type-2 metabotropic glutamate receptor in models of chronic inflammatory, neuropathic pain and visceral nociception. | 28326943 [32] 25378524 [33] |
| DRD3 | chr3 | 114178583 | 114179811 | Ser9Gly polymorphism associated to thermal pain thresholds and noxious inhibitory controls. | 19464960 [34] |
| NR3C1 | chr5 | 143403095 | 143403227 | Chronic stress and early life trauma associated with methylation changes. | 25263804 [35] 26817950 [36] |
| chr5 | 143404021 | 143404121 | |||
| OXT | chr20 | 3071310 | 3071744 | DNA methylation (presumably linked to higher OXT expression) associated to sociability in humans. | 27325757 [37] |
| Mean FM (±St Dev), n = 42 | Mean HCs (±St Dev), n = 42 | t/Chi2 | p-Value | |
|---|---|---|---|---|
| Age (yrs) | 50.359 (±9.685) | 47.564 (±10.576) | −1.4390 | 0.1583 |
| Weight (kg) | 70.459 (±14.017) | 66.203 (±12.742) | −1.7486 | 0.0889 |
| Height (cm) | 161.412 (±5.795) | 161.294 (±6.974) | −0.1086 | 0.9142 |
| BMI (Kg/m2) | 27.413 (±4.772) | 25.675 (±4.860) | −1.803 | 0.0808 |
| Living status (alone, %) | 8 (19.512) | 15 (36.585) | 2.9609 | 0.085 |
| Unemployed (n, %) | 25 (59.523) | 19 (45.238) | 1.718 | 0.190 |
| Number of children | 1.594 (±1.212) | 1.486 (±1.017) | −0.426 | 0.672 |
| Education (n, %) | ||||
| Primary | 26 (61.904) | 19 (46.341) | 3.684 | 0.298 |
| Secondary | 2 (4.878) | 6 (14.634) | ||
| Professional | 7 (16.666) | 6 (14.634) | ||
| University | 7 (16.666) | 10 (24.390) | ||
| Years since diagnosis | 9.027 (96% CI 6.980–11.074) | / | / | / |
| Medications (n, %) | ||||
| Anti-anxiety—bzd | 28 (66.666) | 5 (11.904) | 26.403 | 0.000 |
| Antihistaminic | 3 (7.143) | 3 (7.143) | 0.0000 | 1.000 |
| Gastro-intestinal Cardio-resp | 15 (35.714) | 8 (19.047) | 2.9337 0.0737 | 0.087 0.786 |
| 9 (21.428) | 8 (19.047) | |||
| Antidepressant Antidol/Antinf | 18 (42.857) | 2 (4.762) | 16.800 19.012 | 0.000 0.000 |
| 24 (57.143) | 5 (11.905) | |||
| Antidol/Opioid | 17 (40.476) | 0 (0) | 21.313 | 0.000 |
| BDI | 22.444 (±6.143) | 7.083 (±10.777) | −8.323 | 0.000 |
| BDI cat (no depression) | 26.316 | 81.578 | 23.356 | 0.000 |
| BDI cat (depression) | 73.684 | 18.421 | ||
| PSQI | 13.839 (±4.754) | 7.516 (±5.208) | −6.138 | 0.000 |
| PSQI cat (no sleep impairment) | 48.649 | 85.294 | 10.633 | 0.001 |
| PSQI cat (sleep impairment) | 51.351 | 14.706 | ||
| VAS pain | 7.589 (±1.880) | 1.875 (±1.914) | −12.259 | 0.000 |
| S-FIQ | 67.628 (±17.866) | 16.466 (±15.819) | 11.402 | 0.000 |
| WPI | 13.214 (±3.220) | 2.714 (±1.979) | 18.811 | 0.000 |
| SSS | 9.214 (±1.732) | 3.119 (±2.491) | 13.375 | 0.000 |
| Gene | Chr (Start-Stop) | q-Value | #CpGs | p (MWU) | p (2D KS) | Mean Methylation Level HCs | Mean Methylation Level FM |
|---|---|---|---|---|---|---|---|
| GCSAML | chr1 (247518380–247518621) | 0.083 | 87 | 0 | 0.0069 | 0.15808 | 0.16839 |
| Chr | Start | Stop | q-Value | Mean Methylation Difference | p (MWU) | Mean Methylation Level HCs | Mean Methylation Level FM | |
|---|---|---|---|---|---|---|---|---|
| GCSAML | chr1 | 247518426 | 247518427 | 1 | −0.089641 | 0.019 | 0.42298 | 0.5126 |
| chr1 | 247518439 | 247518440 | 1 | −0.068266 | 0.048 | 0.48173 | 0.55 | |
| chr1 | 247518466 | 247518467 | 1 | −0.070761 | 0.036 | 0.52852 | 0.59929 | |
| chr1 | 247518583 | 247518584 | 0.6 | −0.010065 | 0.00057 | 0.00 | 0.010066 | |
| chr1 | 247518586 | 247518587 | 8.10 × 10−11 | 0.015519 | 7.7 × 10−14 | 0.015519 | 0.00 | |
| DRD3 | chr3 | 114178637 | 114178638 | 1 | −0.035952 | 0.046 | 0.38833 | 0.42429 |
| TRPA1 | chr8 | 72075678 | 72075679 | 1 | −0.017392 | 0.0079 | 0.022132 | 0.039524 |
| chr8 | 72076373 | 72076374 | 1 | −0.024068 | 0.046 | 0.8811 | 0.90517 | |
| chr8 | 72076406 | 72076407 | 1 | −0.016058 | 0.0095 | 1.6667 × 10−7 | 0.016059 | |
| IL25 | chr14 | 23372248 | 23372249 | 1 | −0.038382 | 0.0083 | 0.083523 | 0.1219 |
| OXT | chr20 | 3071336 | 3071337 | 1 | 0.037212 | 0.025 | 0.59888 | 0.56167 |
| chr20 | 3071460 | 3071461 | 1 | −0.010518 | 0.04 | 0.013291 | 0.02381 | |
| chr20 | 3071465 | 3071466 | 1 | −0.015701 | 0.021 | 0.025489 | 0.04119 | |
| chr20 | 3071466 | 3071467 | 1 | −0.014197 | 0.027 | 0.031755 | 0.045952 | |
| chr20 | 3071468 | 3071469 | 1 | 0.012619 | 0.014 | 0.064762 | 0.052143 | |
| MCF2 | chrX | 139692297 | 139692298 | 1 | −0.051717 | 0.0097 | 0.2423 | 0.29402 |
| chrX | 139692312 | 139692313 | 1 | −0.046018 | 0.019 | 0.30653 | 0.35255 |
| Logistic Regression | ||||||
|---|---|---|---|---|---|---|
| Dependent Variable: Fibromyalgia | ||||||
| Variables in the Equation | Odds Ratio | Robust Std. Err. | z | p > |z| | [95% Conf. Interval] | |
| SYT2 | 4.919 | 6.950 | 1.13 | 0.260 | 0.308 | 78.461 |
| GCSAML | 1.038 | 0.162 | 0.24 | 0.808 | 0.765 | 1.410 |
| GRM2 | 0.614 | 0.143 | −2.09 | 0.037 | 0.388 | 0.971 |
| DRD3 | 3.035 | 12.078 | 0.28 | 0.780 | 0.001 | 7413.646 |
| NR3C1 | 0.006 | 0.020 | −1.45 | 0.147 | 5.17 × 10−6 | 6.157 |
| TRPA1 | 12.892 | 22.999 | 1.43 | 0.152 | 0.391 | 425.457 |
| MIR1292 | 0.091 | 0.162 | −1.35 | 0.178 | 0.003 | 2.984 |
| IL25 | 2.279 | 3.888 | 0.48 | 0.629 | 0.080 | 64.565 |
| TNFRSF13B | 7.145 | 15.473 | 0.91 | 0.364 | 0.102 | 498.124 |
| OXT | 1.115 | 0.533 | 0.30 | 0.761 | 0.464 | 2.854 |
| MCF2 | 1.162 | 0.238 | 0.73 | 0.465 | 0.777 | 1.736 |
| Depression—BDI | 1.366 | 0.108 | 3.95 | 0.000 | 1.170 | 1.594 |
| Demographic factors—Cluster Two steps | 2.209 | 2.061 | 0.85 | 0.396 | 0.355 | 13.757 |
| _cons | 3.09 × 10−17 | 8.07 × 10−16 | −1.45 | 0.146 | 1.68 × 10−39 | 567324.4 |
| Number of obs. = 65 | ||||||
| Log pseudolikelihood = −19.74235 | Wald chi2 (7) = 32.48 | |||||
| Prob > chi2 = 0.0020 | ||||||
| Pseudo R2 = 0.5611 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerra, M.C.; Carnevali, D.; Ossola, P.; González-Villar, A.; Pedersen, I.S.; Triñanes, Y.; Donnini, C.; Manfredini, M.; Arendt-Nielsen, L.; Carrillo-de-la-Peña, M.T. DNA Methylation Changes in Fibromyalgia Suggest the Role of the Immune-Inflammatory Response and Central Sensitization. J. Clin. Med. 2021, 10, 4992. https://doi.org/10.3390/jcm10214992
Gerra MC, Carnevali D, Ossola P, González-Villar A, Pedersen IS, Triñanes Y, Donnini C, Manfredini M, Arendt-Nielsen L, Carrillo-de-la-Peña MT. DNA Methylation Changes in Fibromyalgia Suggest the Role of the Immune-Inflammatory Response and Central Sensitization. Journal of Clinical Medicine. 2021; 10(21):4992. https://doi.org/10.3390/jcm10214992
Chicago/Turabian StyleGerra, Maria Carla, Davide Carnevali, Paolo Ossola, Alberto González-Villar, Inge Søkilde Pedersen, Yolanda Triñanes, Claudia Donnini, Matteo Manfredini, Lars Arendt-Nielsen, and Maria Teresa Carrillo-de-la-Peña. 2021. "DNA Methylation Changes in Fibromyalgia Suggest the Role of the Immune-Inflammatory Response and Central Sensitization" Journal of Clinical Medicine 10, no. 21: 4992. https://doi.org/10.3390/jcm10214992