Abstract
Background: Patients with solid cancers and hematopoietic malignancy can experience systemic symptoms compatible with adult-onset Still’s disease (AOSD). The newly described VEXAS, associated with somatic UBA1 mutations, exhibits an overlap of clinical and/or biological pictures with auto inflammatory signs and myelodysplastic syndrome (MDS). Objectives: To describe a cohort of patients with signs of undifferentiated systemic autoinflammatory disorder (USAID) concordant with AOSD and MDS/chronic myelomonocytic leukemia (CMML) and the prevalence of VEXAS proposed management and outcome. Methods: A French multicenter retrospective study from the MINHEMON study group also used for other published works with the support of multidisciplinary and complementary networks of physicians and a control group of 104 MDS/CMML. Results: Twenty-six patients were included with a median age at first signs of USAID of 70.5 years with male predominance (4:1). Five patients met the criteria for confirmed AOSD. The most frequent subtypes were MDS with a blast excess (31%) and MDS with multilineage dysplasia (18%). Seven patients presented with acute myeloid leukemia and twelve died during a median follow-up of 2.5 years. Six out of 18 tested patients displayed a somatic UBA1 mutation concordant with VEXAS, including one woman. High-dose corticosteroids led to a response in 13/16 cases and targeted biological therapy alone or in association in 10/12 patients (anakinra, tocilizumab, and infliximab). Azacytidine resulted in complete or partial response in systemic symptoms for 10/12 (83%) patients including 3 VEXAS. Conclusions: Systemic form of VEXAS syndrome can mimic AOSD. The suspicion of USAID or AOSD in older males with atypia should prompt an evaluation of underlying MDS and assessment of somatic UBA1 mutation.
1. Introduction
Recently, the concept of autoinflammatory disorders keep evolving and an increasing number of new monogenic diseases are identified but numerous autoinflammatory diseases are still to be characterized. Systemic autoinflammatory disorders (SAID) are unprovoked episodic or chronic sterile inflammation which are secondary to innate immune system dysregulation [1]. If well-characterized monogenic diseases such as familial Mediterranean fever are part of it, more than 50% patients presenting with clinical features resembling SAIDs still do not carry any of the know pathogenic mutations in autoinflammatory disease genes [2]. Some patients with SAID fit the criteria for polygenic forms of autoinflammatory diseases, such as adult-onset Still’s disease (AOSD); however, some of them do not. They are defined as undifferentiated systemic autoinflammatory disorders (USAID) [1,3].
Myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML) are part of a heterogeneous group of clonal hematopoietic stem cell diseases characterized by ineffective hematopoiesis, peripheral cytopenia with an increased risk of progression, to acute myeloid leukemia (AML) [4]. About 10 to 20% of MDS/CMML could be associated with various extra hematological features, e.g., neutrophilic dermatoses, systemic vasculitis, inflammatory arthritis, pseudo-Behçet’s disease, and relapsing polychondritis [5,6,7,8]. For years now, the state of autoinflammatory disorders associated with neoplasia reported in the literature were mainly associated with solid cancer but also MDS/CMML, myeloproliferative disorders, and lymphoma, and identified in the literature as “paraneoplastic AOSD” or “malignant associated AOSD” [8,9,10,11,12,13,14,15,16,17].
Recently, VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic), a newly established X-linked disease associated with a somatic mutation in a gene encoding ubiquitin-like modifier activating enzyme 1 (UBA1) characterized by lipid vacuole inclusions in myeloid precursors of bone marrow examination, has been described [18]. These patients often develop hematologic malignancies, and especially MDS.
In this French nationwide study, our objective was to describe patients with MDS/CMML with unexplained recurrent fever and extra hematological signs, concordant or close to AOSD, and fulfilling the definition of USAID, their management, outcomes, overall survival, and the specific risk of progression to AML. We evaluated the prevalence of the newly described VEXAS syndrome in this group of patients and specifically reported their features.
2. Materials and Methods
2.1. Study Design
Data for patients with MDS or CMML and unexplained recurrent fever associated or not to extra hematological signs, concordant with USAID diagnosed between 2010 and June 2020 were retrospectively collected. All patients provided informed consent, and the study followed the Helsinki declaration and received the approval from the Institutional Review Board of Cochin Hospital (CLEP Decision N°: AAA-2021-08040). In agreement with the Committee for the Protection of Individuals—Regional Health Agency of Ile-de-France, given the retrospective nature of this French study, written informed consent from the patients was not required but the non-opposition of patients was necessary for inclusion. Three hundred and ninety patients were included in the MINHEMON registry across France for extra-hematological features associated to MDS/CMML. From this cohort, we selected patients which presented at least fever of unknown origin and elevated phase reactant. Physicians were also asked by Société Nationale Française de Médecine Interne” (SNFMI), “Club Rhumatisme et Inflammation” group (CRI), “Club Médecine INterne, HEMatologie et ONcologie” (MINHEMON) and the “Centre de référence des maladies auto-inflammatoires et des amyloses d’origine inflammatoire” CEREMAIA to report cases of patients with these features associated with MDS or CMML. Inclusion criteria were as follows: (1) MDS or CMML (WHO criteria); (2) AOSD with complete criteria (Yamaguchi and/or Fautrel criteria), or suspicion of AOSD (at least 3 from 5 criteria including fever) consistent with USAID; and (3) an established diagnosis of AOSD and MDS/CMML within the period of 5 years to avoid any fortuitous association. Exclusion criteria was defined as a diagnosis compatible with another autoimmune, infectious, drug-induced or systemic disease at the time of the inclusion. The diagnosis of suspected AOSD or USAID was made based on a consensus by two specialists (MD, AM) and a senior physician from the reference center of autoinflammatory diseases (SG). We compared our patients with a control group of MDS without extra hematological symptoms. Finally, we decided to screen the included patients for the newly described VEXAS to identify if it could be a new phenotype of this disease.
2.2. MDS, CMML
MDS and CMML were diagnosed based on peripheral blood and bone marrow examinations and classified according to the WHO 2016 criteria [19]. Patients were graded using the Revised International Prognostic Scoring System (IPSS-R) [20] for MDS: very low, low, intermediate, high, and very high risk groups; and the new CMML-specific Prognostic Scoring System (CPSS) for CMML: low, intermediate, and high. Karyotypes, the percentage of medullar blasts, and somatic mutations were assessed at the time of MDS diagnosis.
2.3. Genetic Analysis for VEXAS
Somatic UBA1 mutation was explored by incorporating the targeted gene in a homemade 75-gene panel for the next generation sequencing (NGS) of circulating mononucleated cells with a 2% threshold of detection already used in previous work [21]. Only patients alive or dead with available DNA were tested for UBA1. Tested samples were collected in a single center except for one tested in his reference center. UBA1 exon 3 sequences were determined by specific PCR followed by Sanger sequencing reaction using BigDye ™ Terminator (Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s instructions and using the following primers: (forward) 5′-GTGGGTGGGAAAGTCTTTTGT-3′ and (reverse) 5′-TTACAGCTGCCGGGAGTAAAG-3′. A 3500xL-Dx Genetic Analyzer sequencing system was used, and sequences were analyzed with SeqScape software (Applied Biosystems). As MDS patients from the control group did not show any extra hematological features, they were not screened for UBA1 mutation. UBA1 mutated patients were identified and included in French VEXAS group cohort whom data could be used for other studies. The patients used in different studies are represented with an asterisk* in corresponding table.
2.4. USAID
Patient’s clinical files were screened for other autoinflammatory diagnostics. Symptoms of AOSD were specifically recorded and highlighted: fever, skin rash, arthritis or arthralgia, myalgia, pharyngitis or sore throat, nodes, or splenomegaly as well as these severe complications pericarditis, pleuritis, myocarditis, reactive hemophagocytic syndrome (RHS), disseminated intravascular coagulation (DIC), acute respiratory distress syndrome (ARDS), and interstitial lung disease (ILD).
Data from the laboratory test included complete blood count, liver enzymes, C-reactive protein (CRP), ferritin, and glycosylated ferritin levels.
USAID complete clinical and biological response was defined as a total disappearance of all clinical features present at baseline in exclusion of those possible reliable to MDS and/or CMML and a complete normalization of acute-phase reactants. Partial response was defined as an improvement of at least one the clinical sign without an increase of any other features and at least 50% improvement of acute-phase reactants. Patients treated with corticosteroids as first line were differentiated into two groups: high dosage (0.8 to 1 mg/kg/day of prednisone-equivalent) and low dosage. USAID diagnosis was considered concomitant with MDS/CMML or AML when the diagnosis of both diseases was made within ±3 months before or after MDS/CMML were diagnosed. The overall survival, the cause of death and the progression toward AML, were recorded, as well as the date of the last visit.
2.5. Control Group
The control group consisted of 104 patients from the “Groupe Francophone des Myelodysplasies” registry of MDS/CMML which were seen at least once by the internal medicine specialists (AM, OF). These patients did not exhibit any systemic inflammatory or autoimmune features and were selected without matching.
2.6. Ethics
All patients provided informed consent, and the study followed the Helsinki declaration.
2.7. Statistical Analysis
Descriptive data included medians and interquartile ranges (IQR) for continuous variables and frequencies (expressed in percentages) for categorical variables. To account for missing data, results were expressed as observed data (missing data were not replaced). The Chi-square or Fischer’s exact tests were used to compare categorical variables, and the Student’s t-test or Mann–Whitney U test were applied for the comparison of continuous variables. The significance of findings was determined by a p-value of 0.05 or less. Statistical analyses were performed using 2009 GraphPad software, San Diego, CA, USA v6.1.
3. Results
3.1. Characteristics of Patients
All 26 patients with USAID presented with recurrent unexplained fever and elevated acute phase reactants (Figure 1). The median age of appearance of USAID’s symptoms was 70.5 years (IQR: 64.3–78.9) with 76% being male (Table 1). Besides unexplained recurrent fever, clinical features of USAID included arthritis (n = 16, 62%) and skin rash (n = 11, 42%). Severe complications were noted at the time of diagnosis or during the follow-up period in nine patients (35%). These included RHS (n = 5), myopericarditis (n = 3), interstitial lung disease (n = 3), and ARDS (n = 1) (Table 1). Five patients among the initially identified USAID, presented symptoms concordant with a diagnosis of AOSD according to Yamaguchi (n = 5) and Fautrel (n = 4) criteria [22,23]. Nine patients with USAID, reported atypical signs for AOSD including other skin lesions (n = 7, 78%) such as ulcers, nodules, angioedema, erythema nodosum, urticaria, eczematous lesions; non-infectious ileitis (n = 2, 25%), recurrent superficial venous thrombosis (n = 1, 11%), and periorbital headache (n = 1, 11%).
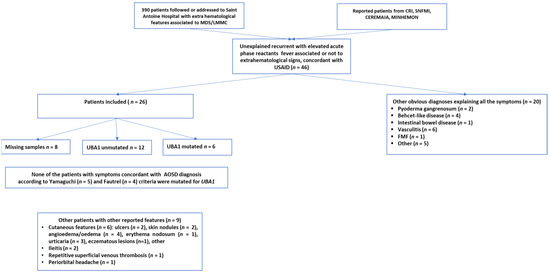
Figure 1.
Flow-chart depicting patient selection. From the 46 patients screened, 26 were included. Six UBA1 mutated patients identified from the included patients.

Table 1.
Baseline characteristics of USAID including pattern suggestive of AOSD.
MDS/CMML characteristics (WHO classification, cytogenetics, and prognostic scores) are shown in Table 2. The most frequent subtype was MDS with excess blasts (30.8% of the patients) (MDS EB-1: 11.5%, MDS EB-2: 19.2%), with a median R-IPSS of 3.7 (IQR: 2–5). NGS somatic mutations screening was available for 17 patients and showed TET2 (n = 5), ASXL1 (n = 4), IDH1 (n = 3), and KRAS (n = 3) mutations (Supplementary Table S1). The diagnosis of USAID co-occurred with preceded and appeared after respectively the diagnosis of MDS/CMML in eight (32%), twelve patients (48%), and five patients (24%). The median time between USAID and MDS/CMML diagnosis was one year (IQR: 0.2–2). Specific hematological treatments were initiated in seventeen cases (65%) within half a year (IQR: 0.2–1.2) after the diagnosis of MDS. First-line treatment mostly consisted of azacytidine (Supplementary Table S2).
Table 2.
Main features of MDS/CMML patients with USAID.
Among the 26 patients included, 18 had samples available to be tested for UBA1 mutation was tested in 18 patients and revealed six 33% positive cases, five out six were males (Figure 1 and Table 3). None of these patients progressed to AML during the 2.2 median years of follow up (ranges 1.2 to 5.6 years). Among these six individuals with VEXAS syndrome, none had a confirmed AOSD, including one with incomplete AOSD criteria sets and five remainders presenting additional unusual clinical features for AOSD. None had polychondritis, one patient hand interstitial pneumonia, one alveolitis, and five had cutaneous features distinct from a rash.
Table 3.
Main features of the patients with UBA1 mutations (VEXAS syndrome).
Compared to MDS/CMML controls without any inflammatory features (n = 104), USAID-associated MDS/CMML patients were younger (71.4 (IQR: 36–85) vs. 78 years (IQR: 42–92); p = 0.0005), without any other statistically significant differences for demography, MDS/CMML subtype, survival. Even if the rates of deaths were higher in the study group (46% vs. 25%; p = 0.02), the overall survival and the time to progression to AML were not significantly different between the groups (Figure 2).
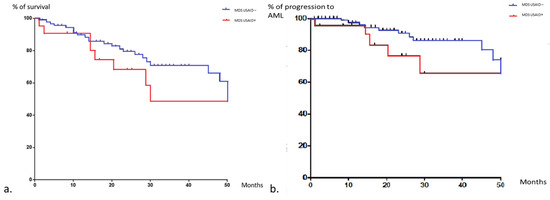
Figure 2.
Kaplan–Meier curves of overall survival (a), progression to AML (b) in MDS patients with and without USAID. Data were censored at time of last visit or death. The response MDS control group is in blue and the one with autoinflammatory features is in red.
3.2. Treatment and Outcomes
Twenty-two patients (85%) received specific treatment for USAID. Corticosteroids were used as a first-line treatment in 19 cases (86%) alone (n = 17) or combined with anakinra or infliximab in the other 2 cases. A complete clinical and biological response to first-line treatment was observed in 14 cases (64%) but 5 cases of secondary steroid dependency were described. Thirteen patients (59%) required a second-line treatment because of a resistance to steroids, primary inefficiency, or relapse, with a response in eight (73%) patients. Targeted biological therapy alone or in association with corticosteroids enabled either a partial or complete response in 9 out of 11 patients (anakinra n = 8/9, tocilizumab n = 0/1, and infliximab n = 1/1). Azacytidine used for MDS or steroid dependent autoinflammatory disease (n = 12) resulted in a complete or partial response of systemic features in 10 (83%) cases. Seven patients (26%) progressed to AML and 12 died (46%), all except one in the VEXAS negative group during the follow-up (Supplementary Table S2 and Figure S1).
Among the six patients with VEXAS, four responded temporarily and/or partially to corticosteroids: relapses and steroid dependency were observed in all cases. Anakinra was used in five cases with a complete or partial response in four cases. Tocilizumab was ineffective in one remaining case. When available, azacytidine led to a response on systemic features in all 3 cases.
4. Discussion
Patients with MDS/CMML can develop USAID with various extra hematological features including pattern suggestive of AOSD. This is the first multidisciplinary series (involving both rheumatologists, hematologists, and clinical immunologists) reporting on USAID associated with MDS/CMML features and the prevalence of VEXAS in such cohort(s). The results of the present study have several important implications. In the case of MDS/CMML with recurrent and unexplained fever, our cohort, although limited, shows that VEXAS syndrome represents 33% (6/18) of cases, these inflammatory situations remaining rare. Moreover, over a median follow-up of 26 month, none of the 6 cases of MDS/CMML VEXAS + progressed to AML, versus 23% of MDS/CMML with USAID and 40% of classic MDS/CMML without inflammatory manifestations. At last, although the numbers are small therefore caution must remain prevalent, there could be a potential interest of biological therapies or even azacytidine when UBA1 is mutated.
Patients with MDS and unexplained recurrent fever should be first tested for the UBA1 mutation. The recent work by Beck et al. which identified VEXAS, implying that somatic mutations may be a more frequent cause of human disease than previously recognized [18,24]. A third of our patients screened for UBA1 were positive for this mutation (including one woman). As a cohort of comparison, in relapsing chondritis, a frequently associated clinical picture usually described with VEXAS, only 7.6% had UBA1 mutations [25]. USAID appeared as a new pattern of VEXAS. Interestingly, 50% of our VEXAS patients experienced RHS. In the future, the order of UBA1 screening could be debated: integrated into the first-line genetic exploration of MDS as well as the MDS/SMP NGS panel or rather screened in the case of MDS/CMML with systemic features in particular USAID.
In contrast to other dysimmune manifestations with a near equal frequency distribution before, simultaneously, and after the diagnosis of MDS/CMML (USAID and systemic features of VEXAS tend to precede the diagnosis of hematological diseases) [9]; hence, the importance of an early identification of warning signs to carefully consider an underlying neoplasia, in particular hematological malignancy. Nearly 20% of the patients matched with the criteria concordant with AOSD, but some atypia should raise the physicians’ attention in patients with newly-diagnosed AOSD, with some features such as late-onset disease [26], male sex [27] as well as cytopenia in particular the absence of leukocytosis (a hallmark feature of AOSD), and macrocytosis and monocytosis [9,11]. In a recent Dutch case series of VEXAS, among patients with unclassified autoinflammation, two among the twelve retrospectively identified were initially suspected with AOSD [28]. Thus, elderly patients with late AOSD diagnosis or suspicion as described in some series [29,30] could also benefit from UBA1 mutation screening.
As described previously for systemic inflammatory and autoimmune disorders associated with MDS/CMML, MDS EB, and MDS MLD were the most frequently associated subtypes in USAID patients not UBA1 mutated [6,31]. No association with a specific karyotype was identified but interestingly TET2 and IDH mutation already associated with systemic inflammatory and auto immune disease and T cell dysregulation in a recent work [32] were also frequently found in our cohort. Two out of the six VEXAS patients presented DNMT3A also described in other VEXAS patients [18,28,33,34], but significance is yet unclear. None of the patients with VEXAS had CMML, which is consistent with the recent report published by Zhao et al. [35]. The prognosis of MDS-associated autoimmune disorders remains unclear, as depending on the types of autoimmune disease in each cohort, the impact on survival could be different [7] but we must notice that in our group, despite a high rate of transformation (more than 25%), the twelve death: only one was UBA1 mutated and none of the VEXAS patients transformed into AML.
The treatment of symptoms of patients with malignant associated features concordant or close to AOSD is challenging. Corticosteroids response was similar to AOSD’s but with a high percentage of steroid dependency or secondary failure [27]. Methotrexate, usually used for its steroid-sparing effect, was rarely applied, presumably due to the risk of cytopenia, explaining the frequent use of biologic agents. The preferential utilization of interleukin-1R antagonists rather than TNF-a antagonists usually used in articular rheumatism is justified by the dominance of the systemic character of the disease with frequent spikes in fever and seemed efficient. Most treatments were only transiently effective, but hematological treatment allowed control of the USAID symptoms for 11 patients in our cohort. As in autoimmune and inflammatory disorders, azacytidine, appears to have a beneficial effect on these systemic manifestations [36,37]. Similarly, in a recent retrospective, Bourbon et al.’s study of the hypomethylating agent and signaling inhibitors seemed to achieve interesting results in VEXAS syndrome patients [38]. The occurrence of USAID or AOSD compatible features over the course of myelodysplastic diseases may prompt a hematological treatment, especially in its refractory form, despite the absence of a direct hematological indication. Thus, azacytidine could be a therapeutical hypothesis for USAID associated with MDS as well as it could be for UBA1 mutated patients.
Our study has several important limitations. First, the retrospective design used here carries an inherited risk of bias and missing data. Second, despite the scarcity of the described clinical syndrome, a small sample size limits the generalizability of the findings reported. Herein we chose to select only patient with occurrence of USAID/AOSD suspicion and MDS/CMML within the period of 5 years to avoid any fortuitous association. Further studies should aim at improving study designs with a prospective nature, larger sample size, and detailed comprehensive information collection.
5. Conclusions
Systemic form of VEXAS syndrome can mimic AOSD, especially in the elderly. USAID symptoms including a pattern suggestive of AOSD associated with MDS/CMML was identified in 33% cases with VEXAS syndrome, thereby further expanding the spectrum of this new syndrome. UBA1 should be sequenced in this population, especially in case of macrocytic associated anemia and chronically elevated CRP among old males, but not exclusively. Although the optimal management of UBA1/MDS overlap patients remains to be determined in larger scale cohorts, biological therapies and/or hypomethylating agents seem promising in this situation.
Key messages:
- Patients with MDS/CMML can develop a recurrent fever with various extra hematological features suggestive of USAID;
- Pseudo-Still disease or USAID associated with a myelodysplastic syndrome are a new phenotype of VEXAS syndrome;
- UBA1 mutation should be screened in case of such association.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/jcm10235586/s1, Figure S1: Different lines of USAID treatments and treatment responses. Twenty-two patient (85%) received specific treatment for USAID and 14 (64%) experienced complete response. Fifty-nine percent of the patient needed a second line treatment and 54% a third line, Table S1: Main features of MDS/CMML patients with USAID, Table S2: USAID including pattern suggestive of AOSD natural history and therapeutical management.
Author Contributions
Conceptualization, M.D. (Marion Delplanque), S.G.-L. and A.M.; Supervision, S.G.-L. and A.M.; Writing—original draft, M.D. (Marion Delplanque); Writing—review & editing, M.D. (Marion Delplanque), A.A., P.H., P.F., J.G., F.M., D.R.-W., N.B., L.D., A.O., M.G., M.M., J.R. (Jerome Razanamahery), G.M., M.D. (Matthieu Décamp), S.M., T.Q., J.R. (Julien Rossignol), L.S., M.S., L.T., A.S., Y.H., S.G.-L. and A.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Cochin Hospital Institutional Review Board (CLEP Decision N°: AAA-2021-08040).
Informed Consent Statement
In agreement with the Committee for the Protection of Individuals—Regional Health Agency of Ile-de-France, given the retrospective nature of this French study, written informed consent from the patients was not required but the non-opposition of patients was necessary for inclusion.
Data Availability Statement
Data are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| A | Arthritis/arthralgia |
| AML | Acute myeloid leukemia |
| ANA | Antinuclear antibody |
| AOSD | Adult-onset Still’s disease |
| ARDS | Acute respiratory distress syndrome |
| C | Cytolysis |
| CBC | Complete blood count |
| CC | Corticosteroids |
| CMML | Chronic myelomonocytic leukemia |
| CPSS | CMML-specific prognostic scoring system |
| CR | Complete remission |
| CRP | C-reactive protein |
| DIC | Disseminated intravascular coagulation |
| F | Female |
| F | Fever |
| Fr | High ferritin |
| G | Low glycosylated ferritin |
| IL | Interleukin |
| ILD | interstitial lung disease |
| L | Lymph nodes and splenomegaly |
| M | Male |
| M/F | Male-to-female ratio |
| MAS | Macrophage activation syndrome |
| MDS | Myelodysplastic syndrome |
| MDS MLD | MDS with multilineage dysplasia |
| MDS SLD | MDS with single lineage dysplasia |
| MDS-EB1 | MDS with excess of blasts <5% |
| MDS-EB2 | MDS with excess of blasts >5% |
| MDS-RS | MDS with ring sideroblasts |
| MM | Myelodysplastic malignancy |
| MTX | Methotrexate |
| PNN | Polymorphonuclear neutrophils >80% |
| PET scan | Positron emission tomography scan |
| PR | Partial remission |
| RF | Rheumatoid factor |
| RHS | Reactive hemophagocytic syndrome |
| R-IPPS | Revised International Prognostic Scoring System |
| SAID | Systemic autoinflammatory disorder |
| S | Skin rash |
| T | Sore throat |
| TNF | Tumor necrosis factor |
| UBA1 | Ubiquitin-like modifier activating enzyme 1 |
| USAID | Undifferentiated systemic autoinflammatory disorder |
| VEXAS | Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic syndrome |
| W | Leukocytosis |
| Yrs | Years |
References
- Papa, R.; Rusmini, M.; Volpi, S.; Caorsi, R.; Picco, P.; Grossi, A.; Caroli, F.; Bovis, F.; Musso, V.; Obici, L.; et al. Next generation sequencing panel in undifferentiated autoinflammatory diseases identifies patients with colchicine-responder recurrent fevers. Rheumatology 2020, 59, 344–360. [Google Scholar] [CrossRef]
- Rusmini, M.; Federici, S.; Caroli, F.; Grossi, A.; Baldi, M.; Obici, L.; Insalaco, A.; Tommasini, A.; Caorsi, R.; Gallo, E.; et al. Next-generation sequencing and its initial applications for molecular diagnosis of systemic auto-inflammatory diseases. Ann. Rheum. Dis. 2016, 75, 1550–1557. [Google Scholar] [CrossRef]
- Harrison, S.R.; McGonagle, D.; Nizam, S.; Jarrett, S.; van der Hilst, J.; McDermott, M.F.; Savic, S. Anakinra as a diagnostic challenge and treatment option for systemic autoinflammatory disorders of undefined etiology. JCI Insight 2016, 1, e86336. [Google Scholar] [CrossRef]
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic syndromes. Lancet 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Fain, O.; Braun, T.; Stirnemann, J.; Fenaux, P. Systemic and autoimmune manifestations in myelodysplastic syndromes. Rev. Med. Interne 2011, 32, 552–559. [Google Scholar] [CrossRef]
- Mekinian, A.; Grignano, E.; Braun, T.; Decaux, O.; Liozon, E.; Costedoat-Chalumeau, N.; Kahn, J.-E.; Hamidou, M.; Park, S.; Puéchal, X.; et al. Systemic inflammatory and autoimmune manifestations associated with myelodysplastic syndromes and chronic myelomonocytic leukaemia: A French multicentre retrospective study. Rheumatology 2016, 55, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Grignano, E.; Jachiet, V.; Fenaux, P.; Ades, L.; Fain, O.; Mekinian, A. Autoimmune manifestations associated with myelodysplastic syndromes. Ann. Hematol. 2018, 97, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, J.K.; Lee, E.Y.; Joo, S.H.; Jung, K.C.; Lee, E.B.; Song, Y.W.; Yoon, S.-S. Certain Autoimmune Manifestations Are Associated with Distinctive Karyotypes and Outcomes in Patients with Myelodysplastic Syndrome: A Retrospective Cohort Study. Medicine 2016, 95, e3091. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, K.; Schett, G.; Manger, B. Adult onset Still’s disease associated with malignancy-Cause or coincidence? Semin. Arthritis Rheum. 2016, 45, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Liozon, E.; Ly, K.H.; Vidal-Cathala, E.; Fauchais, A.-L. Adult-onset Still’s disease as a manifestation of malignancy: Report of a patient with melanoma and literature review. Rev. Med. Interne 2014, 35, 60–64. [Google Scholar] [CrossRef]
- Sun, N.Z.; Brezinski, E.A.; Berliner, J.; Haemel, A.; Connolly, M.K.; Gensler, L.; McCalmont, T.H.; Shinkai, K. Updates in adult-onset Still disease: Atypical cutaneous manifestations and associations with delayed malignancy. J. Am. Acad. Dermatol. 2015, 73, 294–303. [Google Scholar] [CrossRef]
- Sugawara, T.; Tsukada, T.; Wakita, Y.; Wake, Y.; Kouyama, K.; Tamaki, S.; Tanigawa, M.; Iwasaki, E.; Ohta, C.; Kageyama, S. A case of myelodysplastic syndrome progressing to acute myelocytic leukemia in which adult-onset Still’s disease had occurred 6 years before. Int. J. Hematol. 1993, 59, 53–57. [Google Scholar]
- Kacar, M.; Fitton, J.; Gough, A.K.; Buch, M.H.; McGonagle, D.G.; Savic, S. Mixed results with baricitinib in biological-resistant adult-onset Still’s disease and undifferentiated systemic autoinflammatory disease. RMD Open 2020, 6, e001246. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, Y.; Miyamoto, T.; Henzan, H.; Nagafuji, K.; Harada, M.; Akashi, K. Longstanding remission of adult onset Still’s disease under imatinib therapy in a patient with chronic myelogenous leukemia. J. Rheumatol. 2009, 36, 1349–1351. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakagawa, Y.; Furusyo, N.; Taniai, H.; Henzan, H.; Tsuchihashi, T.; Hayashi, J. Chronic myelogenous leukemia that occurred two years after the diagnosis of adult Still’s disease. Intern. Med. 2005, 44, 994–997. [Google Scholar] [CrossRef][Green Version]
- Sono, H.; Matsuo, K.; Miyazato, H.; Sakaguchi, M.; Matsuda, M.; Hamada, K.; Tatsumi, Y.; Maeda, Y.; Funauchi, M.; Kanamaru, A. A case of adult-onset Still’s disease complicated by non-Hodgkin’s lymphoma. Lupus 2000, 9, 468–470. [Google Scholar] [CrossRef]
- Trotta, F.; Dovigo, L.; Scapoli, G.; Cavazzini, L.; Castoldi, G. Immunoblastic malignant lymphoma in adult onset Still’s disease. J. Rheumatol. 1993, 20, 1788–1792. [Google Scholar]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Ospina Cardona, D.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Oganesyan, A.; Jachiet, V.; Chasset, F.; Hirsch, P.; Hage-Sleiman, M.; Fabiani, B.; Duriez, P.; Georgin-Lavialle, S.; Delhommeau, F.; Hakobyan, Y.; et al. VEXAS syndrome: Still expanding the clinical phenotype. Rheumatology 2021, 60, e321–e323. [Google Scholar] [CrossRef]
- Lebrun, D.; Mestrallet, S.; Dehoux, M.; Golmard, J.L.; Granger, B.; Georgin-Lavialle, S.; Arnaud, L.; Grateau, G.; Pouchot, J.; Fautrel, B. Validation of the Fautrel classification criteria for adult-onset Still’s disease. Semin. Arthritis Rheum. 2018, 47, 578–585. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar]
- Levy-Lahad, E.; King, M.-C. Hiding in Plain Sight–Somatic Mutation in Human Disease. N. Engl. J. Med. 2020, 383, 2680–2682. [Google Scholar] [CrossRef]
- Ferrada, M.A.; Sikora, K.A.; Luo, Y.; Wells, K.V.; Patel, B.; Groarke, E.M.; Ospina Cardona, D.; Rominger, E.; Hoffmann, P.; Le, M.T.; et al. Somatic Mutations in UBA1 Define a Distinct Subset of Relapsing Polychondritis Patients with VEXAS Syndrome. Arthritis Rheumatol. 2021, 73, 1886–1895. [Google Scholar] [CrossRef]
- Kalyoncu, U.; Solmaz, D.; Emmungil, H.; Yazici, A.; Kasifoglu, T.; Kimyon, G.; Balkarli, A.; Bes, C.; Ozmen, M.; Alibaz-Oner, F.; et al. Response rate of initial conventional treatments, disease course, and related factors of patients with adult-onset Still’s disease: Data from a large multicenter cohort. J. Autoimmun. 2016, 69, 59–63. [Google Scholar] [CrossRef]
- Giacomelli, R.; Ruscitti, P.; Shoenfeld, Y. A comprehensive review on adult onset Still’s disease. J. Autoimmun. 2018, 93, 24–36. [Google Scholar] [CrossRef]
- van der Made, C.I.; Potjewijd, J.; Hoogstins, A.; Willems, H.P.J.; Kwakernaak, A.J.; de Sevaux, R.G.L.; van Daele, P.L.A.; Simons, A.; Heijstek, M.; Beck, D.B.; et al. Adult-onset autoinflammation caused by somatic mutations in UBA1: A Dutch case series of patients with VEXAS. J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Zoshima, T.; Takeji, A.; Suzuki, Y.; Mizushima, I.; Yamada, K.; Nakashima, A.; Yachie, A.; Kawano, M. Elderly-onset Still’s Disease Complicated by Macrophage Activation Syndrome: A Case Report and Review of the Literature. Intern. Med. 2020, 59, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Mollaeian, A.; Chen, J.; Chan, N.N.; Nizialek, G.A.; Haas, C.J. Adult onset Still’s disease in the elderly: A case-based literature review. BMC Rheumatol. 2021, 5, 12. [Google Scholar]
- de Hollanda, A.; Beucher, A.; Henrion, D.; Ghali, A.; Lavigne, C.; Lévesque, H.; Hamidou, M.; Subra, J.F.; Ifrah, N.; Belizna, C. Systemic and immune manifestations in myelodysplasia: A multicenter retrospective study. Arthritis Care Res. 2011, 63, 1188–1194. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-P.; Boy, M.; Azoulay, C.; Clappier, E.; Sébert, M.; Amable, L.; Klibi, J.; Benlagha, K.; Espeli, M.; Balabanian, K.; et al. MDS/CMML with TET2 or IDH mutation Are Associated with Systemic Inflammatory and Autoimmune Diseases (SIAD) and T Cell Dysregulation. Blood 2020, 136, 31–32. [Google Scholar] [CrossRef]
- Gurnari, C.; Pagliuca, S.; Durkin, L.; Terkawi, L.; Awada, H.; Kongkiatkamon, S.; Zawit, M.; Hsi, E.D.; Carraway, H.E.; Rogers, H.J.; et al. Vacuolization of hematopoietic precursors: An enigma with multiple etiologies. Blood 2021, 137, 3685–3689. [Google Scholar] [CrossRef] [PubMed]
- Obiorah, I.E.; Patel, B.A.; Groarke, E.M.; Wang, W.; Trick, M.; Ombrello, A.K.; Ferrada, M.A.; Wu, Z.; Gutierrez-Rodrigues, F.; Lotter, J.; et al. Benign and malignant hematologic manifestations in patients with VEXAS syndrome due to somatic mutations in UBA1. Blood Adv. 2021, 5, 3203–3215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-P.; Schell, B.; Sébert, M.; Kim, R.; Lemaire, P.; Boy, M.; Mathis, S.; Larcher, L.; Chauvel, C.; Dhouaieb, M.B.; et al. Prevalence of UBA1 mutations in MDS/CMML patients with systemic inflammatory and auto-immune disease. Leukemia 2021, 35, 2731–2733. [Google Scholar] [CrossRef]
- Fraison, J.-B.; Mekinian, A.; Grignano, E.; Kahn, J.-E.; Arlet, J.-B.; Decaux, O.; Denis, G.; Buchdahl, A.-L.; Omouri, M.; Maigne, G.; et al. Efficacy of Azacitidine in autoimmune and inflammatory disorders associated with myelodysplastic syndromes and chronic myelomonocytic leukemia. Leuk. Res. 2016, 43, 13–17. [Google Scholar] [CrossRef]
- Frietsch, J.J.; Dornaus, S.; Neumann, T.; Scholl, S.; Schmidt, V.; Kunert, C.; Sayer, H.G.; Hochhaus, A.; La Rosée, P. Paraneoplastic inflammation in myelodysplastic syndrome or bone marrow failure: Case series with focus on 5-azacytidine and literature review. Eur. J. Haematol. 2014, 93, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Bourbon, E.; Heiblig, M.; Gerfaud Valentin, M.; Barba, T.; Durel, C.-A.; Lega, J.C.; Barraco, F.; Sève, P.; Jamilloux, Y.; Sujobert, P. Therapeutic options in VEXAS syndrome: Insights from a retrospective series. Blood 2021, 137, 3682–3684. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).