
Extramedullary Hematopoiesis of the Liver and Spleen


 , , , , and
, , , , and
Abstract
:1. Introduction
2. The Bone Marrow Hematopoietic Niche
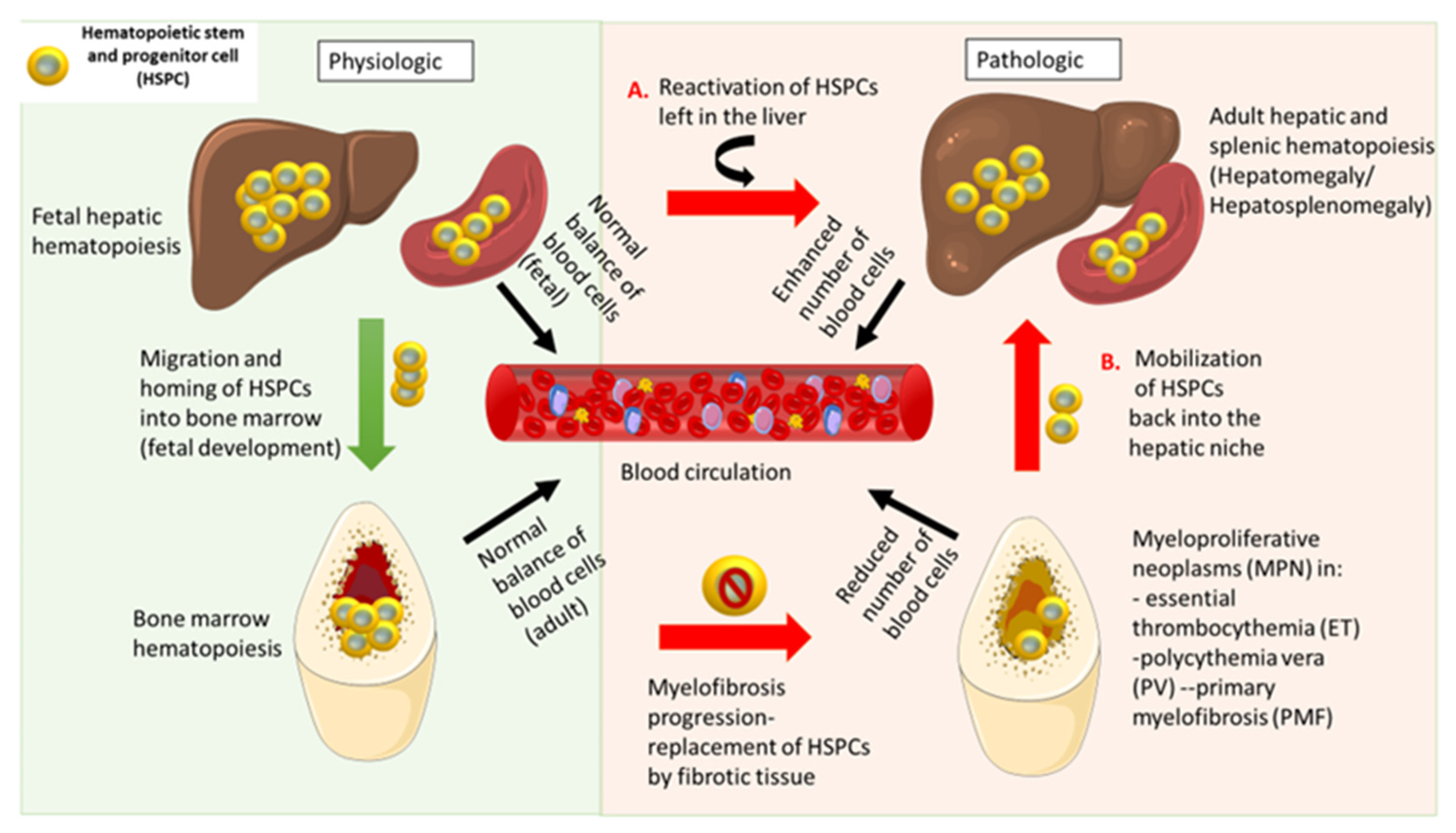
3. Extramedullary Hematopoiesis

{kind=link}
| Pathology | Sites of EMH | Lineage | Number of Cases | Human/Animal Studies | References |
|---|---|---|---|---|---|
| Primary myelofibrosis | Spleen liver | 5 patients | Human | [22] | |
| Secondary myelofibrosis after essential thrombocythemia | Spleen | 1 patient | Human | [22] | |
| Primary myelofibrosis | Spleen | Malignant primitive HPCs (PB CD34+) | 8 patients | Human | [23] |
| Allograft liver transplant | Liver | 27 patients | [30] | ||
| Acute promyelocytic leukemia (APL) | Central nervous system, skin, lung, | Leukemia cells | 21 patients | Human | [31] |
| Chronic myelogenous leukemia | Stomach | Case reports | Human | [3,4,32] | |
| Thalassemia | N/A | 10 patients | Human | [33] | |
| Agnogenic myeloid metaplasia | Lung | 2 | Human | [34] | |
| Primary myelofibrosis (MMM) | Spleen | Granulocyte infiltration | 213 patients (MMM) | Human | [35] |
| Myelofibrosis with myeloid metaplasia (MMM) with thalidomide therapy | Liver/spleen Pericardium | Red blood cells Granulocytic precursors | 1 of 7 patients | Human | [36] |
| Chronic myeloproliferative diseases | Spleen | JAK2(V617F) | 15 patients 47 patients | Human Human | [37] [38] |
| Experimental spondylarthritis | Spleen Inflamed joints | Myelopoiesis | 39 mice (13 mice/group) | Animal/experimental | [25] |
| Inflammation | Spleen, liver, other sites | Megakaryopoiesis | N/A | Animal | [28] |
| Experimental mutations in the SETD1B gene; | Spleen | Myeloid and lymphoid | N/A | Animal | [39] |
| Acute myeloid leukemia | Spleen | N/A | Animal | [40] | |
| Experimental overexpression of Tlx1 | Spleen | N/A | Animal | [29] | |
| Experimental Dnmt3a loss of function | Spleen, liver | HSC | 18 mice | Animal | [41] |
4. Pathological Hematopoiesis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Notta, F.; Zandi, S.; Takayama, N.; Dobson, S.; Gan, O.I.; Wilson, G.; Kaufmann, K.B.; McLeod, J.; Laurenti, E.; Dunant, C.F.; et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 2016, 351, aab2116. [Google Scholar] [CrossRef] [Green Version]
- McGrath, K.E.; Frame, J.M.; Palis, J. Early hematopoiesis and macrophage development. Semin. Immunol. 2015, 27, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Cognein, P.; Belgrano, V.; Mastracci, L.; Pitto, F.; Fasoli, A.; De Cian, F.; Grillo, F. Endoscopic management of a rare cause of upper gastrointestinal bleeding: Gastric polypoid extramedullary hemopoiesis. Endoscopy 2014, 46, E674–E675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, G.M.; Shortsleeve, M.J. Gastric polyps due to extramedullary hematopoiesis. AJR Am. J. Roentgenol. 1998, 171, 531. [Google Scholar] [CrossRef]
- Popescu, D.-M.; Botting, R.A.; Stephenson, E.; Green, K.; Webb, S.; Jardine, L.; Calderbank, E.F.; Polański, K.; Goh, I.; Efremova, M.; et al. Decoding human fetal liver haematopoiesis. Nature 2019, 574, 365–371. [Google Scholar] [CrossRef]
- Qian, L.; Krause, D.S.; Saltzman, W.M. Enhanced growth and hepatic differentiation of fetal liver epithelial cells through combinational and temporal adjustment of soluble factors. Biotechnol. J. 2012, 7, 440–448. [Google Scholar] [CrossRef]
- Martin, M.A.; Bhatia, M. Analysis of the human fetal liver hematopoietic microenvironment. Stem Cells Dev. 2005, 14, 493–504. [Google Scholar] [CrossRef]
- Freedman, M.H.; Saunders, E.F. Hematopoiesis in the human spleen. Am. J. Hematol. 1981, 11, 271–275. [Google Scholar] [CrossRef]
- Ivanovs, A.; Rybtsov, S.; Ng, E.S.; Stanley, E.G.; Elefanty, A.G.; Medvinsky, A. Human haematopoietic stem cell development: From the embryo to the dish. Development 2017, 144, 2323–2337. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Chen, D.; Long, H.; Zhu, B. The mechanisms of pathological extramedullary hematopoiesis in diseases. Cell. Mol. Life Sci. 2020, 77, 27–38. [Google Scholar] [CrossRef]
- Crane, G.M.; Jeffery, E.; Morrison, S.J. Adult haematopoietic stem cell niches. Nat. Rev. Immunol. 2017, 17, 573–590. [Google Scholar] [CrossRef] [PubMed]
- Chagraoui, J.; Lepage-Noll, A.; Anjo, A.; Uzan, G.; Charbord, P. Fetal liver stroma consists of cells in epithelial-to-mesenchymal transition. Blood 2003, 101, 2973–2982. [Google Scholar] [CrossRef] [Green Version]
- Laurenti, E.; Göttgens, B. From haematopoietic stem cells to complex differentiation landscapes. Nature 2018, 553, 418–426. [Google Scholar] [CrossRef]
- Spangrude, G.J.; Heimfeld, S.; Weissman, I.L. Purification and characterization of mouse hematopoietic stem cells. Science 1988, 241, 58–62. [Google Scholar] [CrossRef]
- Satterthwaite, A.B.; Burn, T.C.; Le Beau, M.M.; Tenen, D.G. Structure of the gene encoding CDa human hematopoietic stem cell antigen. Genomics 1992, 12, 788–794. [Google Scholar] [CrossRef]
- Kapatia, G.; Kaur, A.; Rastogi, P.; Sreedharanunni, S.; Gupta, P.; Rohilla, M.; Gupta, N.; Srinivasan, R.; Rajwanshi, A.; Dey, P. Extramedullary hematopoiesis: Clinical and cytological features. Diagn Cytopathol. 2020, 48, 191–196. [Google Scholar] [CrossRef]
- Hu, D.; Shilatifard, A. Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 2016, 34, 2021–2041. [Google Scholar] [CrossRef]
- Li, L.; Duan, M.; Chen, W.; Jiang, A.; Li, X.; Yang, J.; Li, Z. The spleen in liver cirrhosis: Revisiting an old enemy with novel targets. J. Transl. Med. 2017, 15, 111. [Google Scholar] [CrossRef] [Green Version]
- De Leo, E.K.; Shah, C.P.; Grajo, J.R.; Liu, X.; Parekh, H. Extramedullary Hematopoiesis in Mismatch Repair Deficient Colon Cancer Patient on Adjuvant Chemotherapy. Cureus 2021, 13, e12899. [Google Scholar] [CrossRef]
- Tarantino, G.; Scalera, A.; Finelli, C. Liver-spleen axis: Intersection between immunity, infections and metabolism. World J. Gastroenterol. 2013, 2021, 3534–3542. [Google Scholar] [CrossRef]
- Wardemann, H.; Boehm, T.; Dear, N.; Carsetti, R. B-1a B cells that link the innate and adaptive immune responses are lacking in the absence of the spleen. J. Exp. Med. 2002, 195, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Uribe, M.; Morel, O.; Ungureanu, C.; Desterke, C.; Le Bousse-Kerdilès, M.C.; Boulahdour, H. Assessment of sites of marrow and extramedullary hematopoiesis by hybrid imaging in primary myelofibrosis patients. Cancer Med. 2016, 5, 2378–2384. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Prakash, S.; Lu, M.; Tripodi, J.; Ye, F.; Najfeld, V.; Li, Y.; Schwartz, M.; Weinberg, R.; Roda, P.; et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J. Clin. Investig. 2012, 122, 3888–3899. [Google Scholar] [CrossRef]
- Fan, N.; Lavu, S.; Hanson, C.A.; Tefferi, A. Extramedullary hematopoiesis in the absence of myeloproliferative neoplasm: Mayo Clinic case series of 309 patients. Blood Cancer J. 2018, 8, 1–4. [Google Scholar] [CrossRef]
- Regan-Komito, D.; Swann, J.W.; Demetriou, P.; Cohen, E.S.; Horwood, N.J.; Sansom, S.N.; Griseri, T. GM-CSF drives dysregulated hematopoietic stem cell activity and pathogenic extramedullary myelopoiesis in experimental spondyloarthritis. Nat. Commun. 2020, 11, 155. [Google Scholar] [CrossRef]
- Fernández-García, V.; González-Ramos, S.; Martín-Sanz, P.; Castrillo, A.; Boscá, L. Contribution of Extramedullary Hematopoiesis to Atherosclerosis. The Spleen as a Neglected Hub of Inflammatory Cells. Front. Immunol. 2020, 11, 2790. [Google Scholar] [CrossRef]
- Huang, Z.; Richmond, T.D.; Muntean, A.G.; Barber, D.L.; Weiss, M.J.; Crispino, J.D. STAT1 promotes megakaryopoiesis downstream of GATA-1 in mice. J. Clin. Investig. 2007, 117, 3890–3899. [Google Scholar] [CrossRef] [PubMed]
- Haas, S.; Hansson, J.; Klimmeck, D.; Loeffler, D.; Velten, L.; Uckelmann, H.; Wurzer, S.; Prendergast, Á.M.; Schnell, A.; Hexel, K.; et al. Inflammation-Induced Emergency Megakaryopoiesis Driven by Hematopoietic Stem Cell-like Megakaryocyte Progenitors. Cell Stem Cell 2015, 17, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Oda, A.; Tezuka, T.; Ueno, Y.; Hosoda, S.; Amemiya, Y.; Notsu, C.; Kasahara, T.; Nishiyama, C.; Goitsuka, R. Niche-induced extramedullary hematopoiesis in the spleen is regulated by the transcription factor Tlx1. Sci. Rep. 2018, 8, 8308. [Google Scholar] [CrossRef] [Green Version]
- Tsamandas, A.C.; Jain, A.B.; Raikow, R.B.; Demetris, A.J.; Nalesnik, M.A.; Randhawa, P.S. Extramedullary hematopoiesis in the allograft liver. Mod. Pathol. 1995, 8, 671–674. Available online: https://pubmed.ncbi.nlm.nih.gov/8532704/ (accessed on 15 October 2021).
- Specchia, G.; Coco, F.L.; Vignetti, M.; Avvisati, G.; Fazi, P.; Albano, F.; Di Raimondo, F.; Martino, B.; Ferrara, F.; Selleri, C.; et al. Extramedullary involvement at relapse in acute promyelocytic leukemia patients treated or not with all-trans retinoic acid: A report by the Gruppo Italiano Malattie Ematologiche dell’Adulto. J. Clin. Oncol. 2001, 19, 4023–4028. [Google Scholar] [CrossRef]
- Gomes, A.S.; Harell, G.S. Tumefactive extramedullary hematopoiesis of the stomach. Gastrointest. Radiol. 1976, 1, 163–165. [Google Scholar] [CrossRef]
- Teawtrakul, N.; Jetsrisuparb, A.; Pongudom, S.; Sirijerachai, C.; Chansung, K.; Wanitpongpun, C.; Fucharoen, S. Epidemiologic study of major complications in adolescent and adult patients with thalassemia in Northeastern Thailand: The E-SAAN study phase I. Hematology 2018, 23, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Asakura, S.; Colby, T.V. Agnogenic myeloid metaplasia with extramedullary hematopoiesis and fibrosis in the lung: Report of two cases. Chest 1994, 105, 1866–1868. [Google Scholar] [CrossRef] [Green Version]
- Mesa, R.A.; Li, C.Y.; Schroeder, G.; Tefferi, A. Clinical correlates of splenic histopathology and splenic karyotype in myelofibrosis with myeloid metaplasia. Blood 2001, 97, 3665–3667. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.A.; Mesa, R.A.; Li, C.-Y.; Hook, C.C.; Ansell, S.M.; Levitt, R.M.; Geyer, S.M.; Tefferi, A. Thalidomide treatment in myelofibrosis with myeloid metaplasia. Br. J. Haematol. 2002, 117, 288–296. [Google Scholar] [CrossRef]
- Konoplev, S.; Hsieh, P.P.; Chang, C.C.; Medeiros, L.J.; Lin, P. Janus kinase 2 V617F mutation is detectable in spleen of patients with chronic myeloproliferative diseases suggesting a malignant nature of splenic extramedullary hematopoiesis. Hum. Pathol. 2007, 38, 1760–1763. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.-P.; Olsen, R.J.; O’Malley, D.P.; Konoplev, S.N.; Hussong, J.W.; Dunphy, C.H.; Perkins, S.L.; Cheng, L.; Lin, P.; Chang, C.-C. The role of Janus Kinase 2 V617F mutation in extramedullary hematopoiesis of the spleen in neoplastic myeloid disorders. Mod. Pathol. 2007, 20, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Zhang, Q.; Tasdogan, A.; Petzold, A.; Dahl, A.; Arneth, B.M.; Slany, R.; Fehling, H.J.; Kranz, A.; Stewart, A.F.; et al. The H3K4 methyltransferase setd1b is essential for hematopoietic stem and progenitor cell homeostasis in mice. eLife 2018, 7, e27157. [Google Scholar] [CrossRef]
- Pandey, R.; Ramdas, B.; Wan, C.; Sandusky, G.; Mohseni, M.; Zhang, C.; Kapur, R. SHP2 inhibition reduces leukemogenesis in models of combined genetic and epigenetic mutations. J. Clin. Investig. 2019, 8, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Mayle, A.; Yang, L.; Rodriguez, B.; Zhou, T.; Chang, E.; Curry, C.V.; Challen, G.A.; Li, W.; Wheeler, D.; Rebel, V.I.; et al. Hematopoiesis and stem cells: Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015, 125, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Božić, T.; Frobel, J.; Raic, A.; Ticconi, F.; Kuo, C.C.; Heilmann-Heimbach, S.; Goecke, T.W.; Zenke, M.; Jost, E.; Costa, I.G.; et al. Variants of DNMT3A cause transcript-specific DNA methylation patterns and affect hematopoiesis. Life Sci. Alliance 2018, 1, e201800153. [Google Scholar] [CrossRef] [Green Version]
- Ponciano-Gómez, A.; Martínez-Tovar, A.; Vela-Ojeda, J.; Olarte-Carrillo, I.; Centeno-Cruz, F.; Garrido, E. Mutations in TET2 and DNMT3A genes are associated with changes in global and gene-specific methylation in acute myeloid leukemia. Tumor Biol. 2017, 39, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Celik, H.; Kramer, A.; Challen, G.A. DNA methylation in normal and malignant hematopoiesis. Int. J. Hematol. 2016, 103, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulei, D.; Tomuleasa, C.; Qian, L.; Bagacean, C.; Croce, C.M.; Ghiaur, G. Editorial: Novel Drugs Targeting the Microenvironment and the Epigenetic Changes in Hematopoietic Malignancies. Front. Pharmacol. 2020, 11, 614614. [Google Scholar] [CrossRef]
- Goyama, S.; Kitamura, T. Epigenetics in normal and malignant hematopoiesis: An overview and update 2017. Cancer Sci. 2017, 108, 553–562. [Google Scholar] [CrossRef] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.; Schuh, A.C.; Yee, K.W.; et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef]
- Langstein, J.; Milsom, M.D.; Lipka, D.B. Impact of DNA methylation programming on normal and pre-leukemic hematopoiesis. Semin. Cancer Biol. 2018, 51, 89–100. [Google Scholar] [CrossRef]
- Thapa, B.; Fazal, S.; Parsi, M.; Rogers, H.J. Myeloproliferative Neoplasms; StatPearls Publishing: Washington, DC, USA, 2020. [Google Scholar]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef]
- Kiladjian, J.J. The spectrum of JAK2-positive myeloproliferative neoplasms. Hematol. Am. Soc. Hematol. Educ. Progr. 2012, 2012, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Muxí, P.J.; Oliver, A.C. Jak-2 positive myeloproliferative neoplasms. Curr. Treat. Options Oncol. 2014, 15, 147–156. [Google Scholar] [CrossRef]
- Poluben, L.; Puligandla, M.; Neuberg, D.; Bryke, C.R.; Hsu, Y.; Shumeiko, O.; Yuan, X.; Voznesensky, O.; Pihan, G.; Adam, M.; et al. Characteristics of myeloproliferative neoplasms in patients exposed to ionizing radiation following the Chernobyl nuclear accident. Am. J. Hematol. 2019, 94, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Gross-Davis, C.A.; Heavner, K.; Frank, A.L.; Newschaffer, C.; Klotz, J.; Santella, R.M.; Burstyn, I. The role of genotypes that modify the toxicity of chemical mutagens in the risk for myeloproliferative neoplasms. Int. J. Environ. Res. Public Health 2015, 12, 2465–2485. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Haider, M.Z.; Anwer, F. Genetics, Philadelphia Chromosome; StatPearls Publishing: New York, NY, USA, 2021. [Google Scholar]
- Soverini, S.; Bassan, R.; Lion, T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Liu-Dumlao, T.; Kantarjian, H.; Thomas, D.A.; O’Brien, S.; Ravandi, F. Philadelphia-positive acute lymphoblastic leukemia: Current treatment options. Curr. Oncol. Rep. 2012, 14, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Yildiz, I.; Yokuş, O.; Gedik, H. Janus kinase 2 mutations in cases with BCR-ABL-negative chronic myeloproliferative disorders from Turkey. Avicenna J. Med. 2017, 7, 28. [Google Scholar] [CrossRef]
- Ojeda, M.J.; Bragós, I.M.; Calvo, K.L.; Williams, G.M.; Carbonell, M.M.; Pratti, A.F. CALR, JAK2 and MPL mutation status in Argentinean patients with BCR-ABL1- negative myeloproliferative neoplasms. Hematology 2018, 23, 208–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Thiele, J.; Vannucchi, A.M.; Barbui, T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014, 28, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- McKerrell, T.; Park, N.; Chi, J.; Collord, G.; Moreno, T.; Ponstingl, H.; Dias, J.; Gerasimou, P.; Melanthiou, K.; Prokopiou, C.; et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Advances. Am. Soc. Hematol. 2017, 1, 968–971. [Google Scholar]
- Plo, I.; Bellanné-Chantelot, C.; Mosca, M.; Mazzi, S.; Marty, C.; Vainchenker, W. Genetic alterations of the thrombopoietin/MPL/JAK2 axis impacting Megakaryopoiesis. Front. Endocrinol. 2017, 8, 234. [Google Scholar] [CrossRef]
- Akpinar, T.S.; Hançer, V.S.; Nalçaci, M.; Diz-Küçükkaya, R. Kronik miyeloproliferatif neoplazmlarda MPL W515L/K mutasyonlari. Turkish J. Hematol. 2013, 30, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood. Am. Soc. Hematol. 2017, 129, 1607–1616. [Google Scholar]
- Shide, K.; Kameda, T.; Kamiunten, A.; Ozono, Y.; Tahira, Y.; Yokomizo-Nakano, T.; Kubota, S.; Ono, M.; Lkeda, K.; Sekine, M.; et al. Calreticulin haploinsufficiency augments stem cell activity and is required for onset of myeloproliferative neoplasms in mice. Blood 2020, 136, 106–118. [Google Scholar] [PubMed]
- Saeidi, K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit. Rev. Oncol. 2016, 98, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.J.; Roda, P.I.; Dorion, P. Molecular characterization of a transformation from primary myelofibrosis into polycythemia vera: A case report. Blood 2013, 122, 297–298. [Google Scholar] [CrossRef] [Green Version]
- Harrison, C.N.; Koschmieder, S.; Foltz, L.; Guglielmelli, P.; Flindt, T.; Koehler, M.; Mathias, J.; Komatsu, N.; Boothroyd, R.N.; Spierer, A.; et al. The impact of myeloproliferative neoplasms (MPNs) on patient quality of life and productivity: Results from the international MPN Landmark survey. Ann. Hematol. 2017, 96, 1653–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carobbio, A.; Ferrari, A.; Masciulli, A.; Ghirardi, A.; Barosi, G.; Barbui, T. Leukocytosis and thrombosis in essential thrombocythemia and polycythemia vera: A systematic review and meta-analysis. Blood Adv. 2019, 3, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Fowlkes, S.; Murray, C.; Fulford, A.; De Gelder, T.; Siddiq, N. Myeloproliferative neoplasms (MPNs)—Part 1: An overview of the diagnosis and treatment of the “classical” MPNs. Can. Oncol. Nurs. J. 2018, 28, 262–268. [Google Scholar] [CrossRef]
- Chapman, J.; Bansal, P.; Goyal, A.; Azevedo, A.M. Splenomegaly. The 5-Minute Pediatric Consult, 8th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2021; pp. 870–871. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Hematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cenariu, D.; Iluta, S.; Zimta, A.-A.; Petrushev, B.; Qian, L.; Dirzu, N.; Tomuleasa, C.; Bumbea, H.; Zaharie, F. Extramedullary Hematopoiesis of the Liver and Spleen. J. Clin. Med. 2021, 10, 5831. https://doi.org/10.3390/jcm10245831
Cenariu D, Iluta S, Zimta A-A, Petrushev B, Qian L, Dirzu N, Tomuleasa C, Bumbea H, Zaharie F. Extramedullary Hematopoiesis of the Liver and Spleen. Journal of Clinical Medicine. 2021; 10(24):5831. https://doi.org/10.3390/jcm10245831
Chicago/Turabian StyleCenariu, Diana, Sabina Iluta, Alina-Andreea Zimta, Bobe Petrushev, Liren Qian, Noemi Dirzu, Ciprian Tomuleasa, Horia Bumbea, and Florin Zaharie. 2021. "Extramedullary Hematopoiesis of the Liver and Spleen" Journal of Clinical Medicine 10, no. 24: 5831. https://doi.org/10.3390/jcm10245831
APA StyleCenariu, D., Iluta, S., Zimta, A.-A., Petrushev, B., Qian, L., Dirzu, N., Tomuleasa, C., Bumbea, H., & Zaharie, F. (2021). Extramedullary Hematopoiesis of the Liver and Spleen. Journal of Clinical Medicine, 10(24), 5831. https://doi.org/10.3390/jcm10245831