Linking Labile Heme with Thrombosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
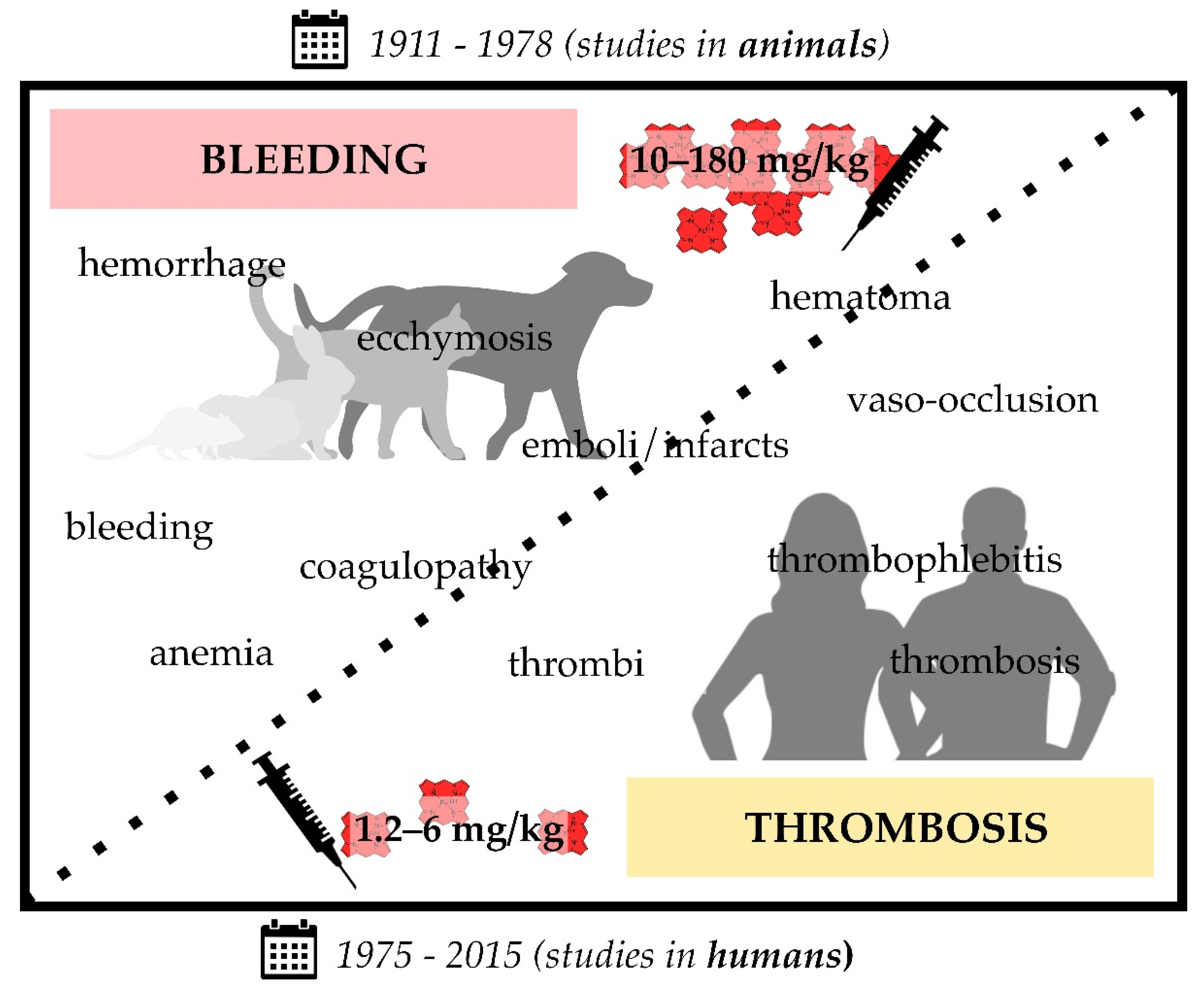
2. Thrombotic Complications upon Heme Injection
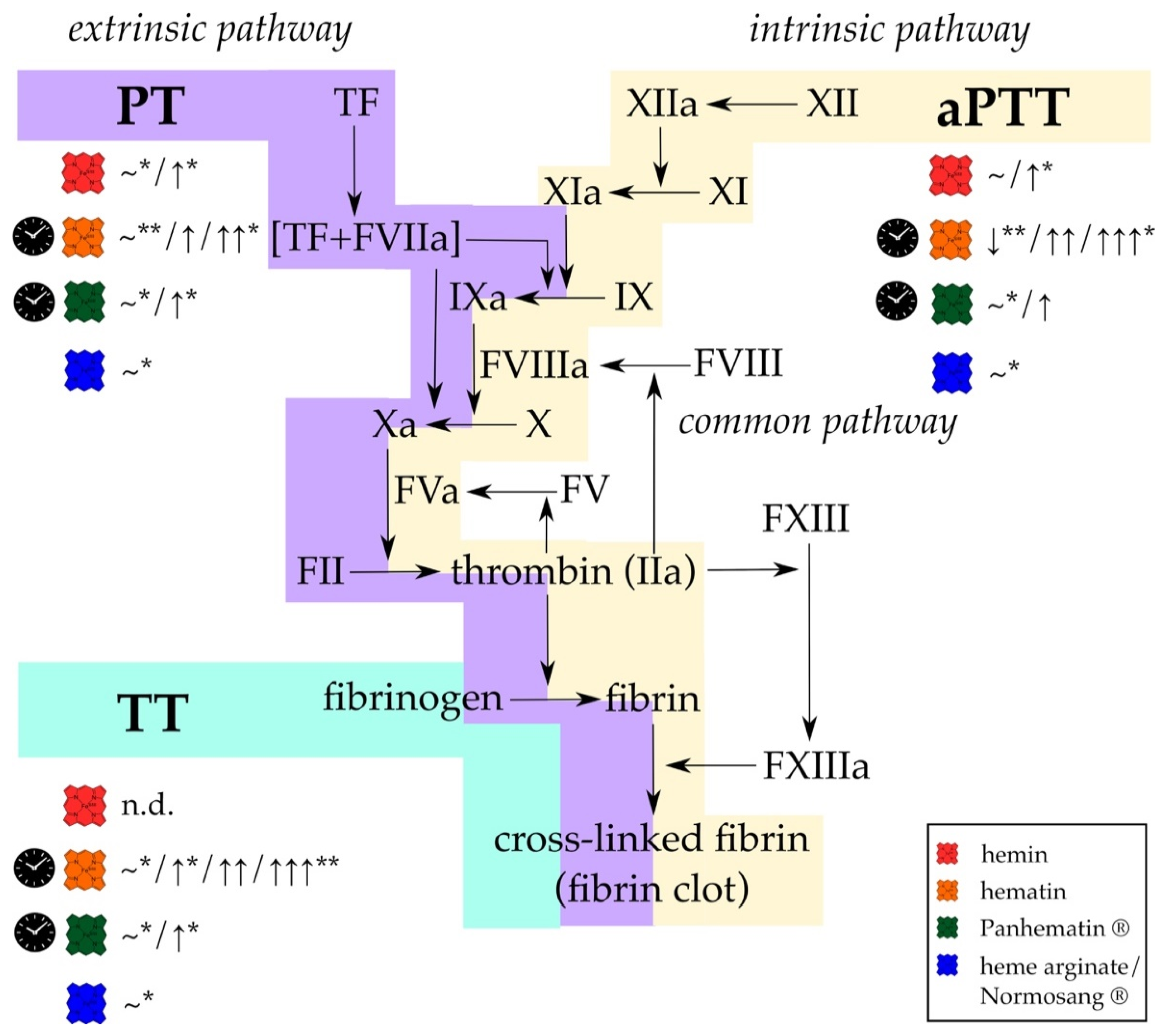
3. Heme-Mediated Interference in Coagulation Point-of-Care Testing
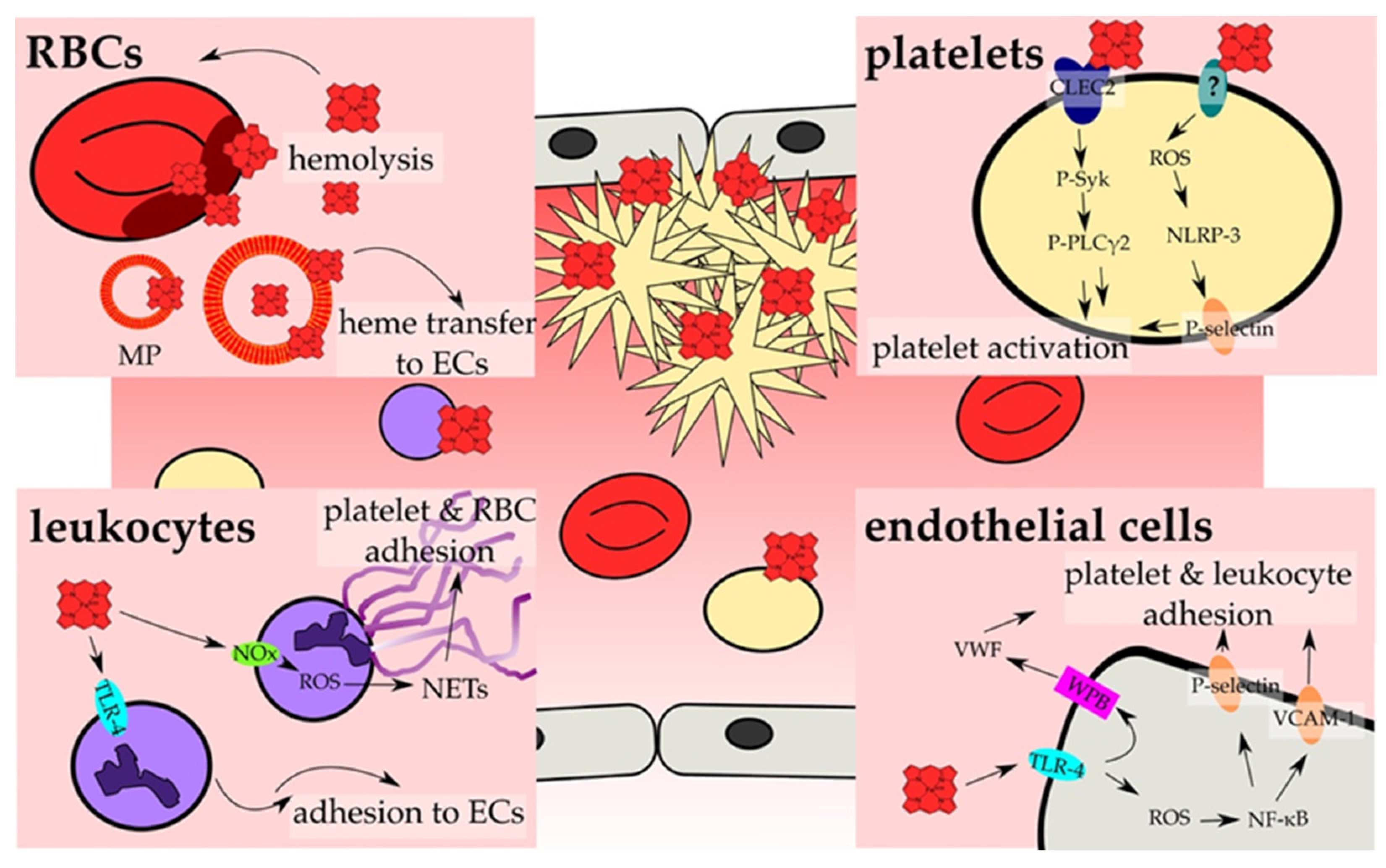
4. Heme Promotes Clotting Processes by Affecting Involved Cells
4.1. Heme and Platelets
4.2. Heme and Endothelial Cells
4.3. Heme and RBCs
4.4. Heme and Leukocytes
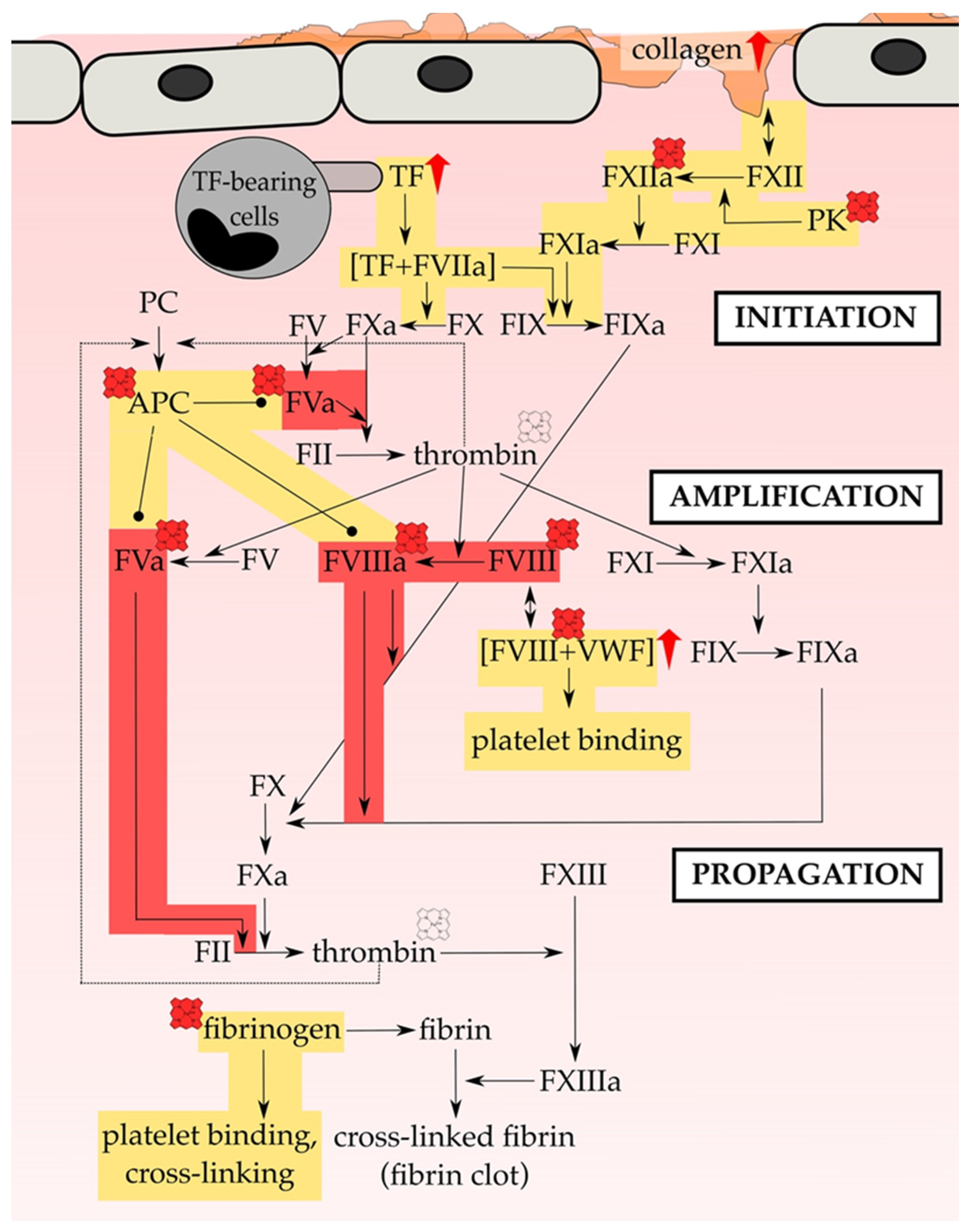
5. Direct Influence of Heme on Coagulation Proteins
5.1. Heme Interaction with Fibrin(ogen)
5.2. Heme Interaction with Factor VIIIa (FVIII(a)) and VWF
5.3. Heme Interaction with Factor V (FV)
5.4. Heme Interaction with Factor XII (FXII)
5.5. Heme Interaction with Thrombin
5.6. Heme Interaction with Plasmin(Ogen)
5.7. Heme Interaction with Adhesion Proteins
5.8. Upregulation of TF by Heme
5.9. Heme Interaction with Anticoagulant Proteins
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raskob, G.E.; Angchaisuksiri, P.; Blanco, A.N.; Buller, H.; Gallus, A.; Hunt, B.J.; Hylek, E.M.; Kakkar, A.; Konstantinides, S.V.; McCumber, M.; et al. Thrombosis: A major contributor to global disease burden. Thromb. Res. 2014, 134, 931–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008, 359, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Oklu, R. Thrombosis. Cardiovasc. Diagn. Ther. 2017, 7, S131–S133. [Google Scholar] [CrossRef]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2016, 38, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Prandoni, P. Venous and arterial thrombosis: Two aspects of the same disease? Clin. Epidemiol. 2009, 1, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Previtali, E.; Bucciarelli, P.; Passamonti, S.M.; Martinelli, I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011, 9, 120–138. [Google Scholar]
- Froemel, D.; Fitzsimons, S.-J.; Frank, J.; Sauerbier, M.; Meurer, A.; Barker, J.H. A review of thrombosis and antithrombotic therapy in microvascular surgery. Eur. Surg. Res. 2013, 50, 32–43. [Google Scholar] [CrossRef]
- Knudson, M.M.; Ikossi, D.G.; Khaw, L.; Morabito, D.; Speetzen, L.S. Thromboembolism after trauma. Ann. Surg. 2004, 240, 490–498. [Google Scholar] [CrossRef]
- Wilkerson, W.R.; Sane, D.C. Aging and thrombosis. Semin. Thromb. Hemost. 2002, 28, 555–568. [Google Scholar] [CrossRef]
- Greinacher, A.; Pötzsch, B. Coagulation/Thrombosis. In Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer Berlin Heidelberg: Berlin, Germany, 2008; pp. 375–380. [Google Scholar]
- Green, D.; Scott, J.P. Is sickle cell crisis a thrombotic event? Am. J. Hematol. 1986, 23, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.; Pawlinski, R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br. J. Haematol. 2013, 162, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillery, C.A.; Panepinto, J.A. Pathophysiology of stroke in sickle cell disease. Microcirculation 2004, 11, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Ataga, K.I.; Cappellini, M.D.; Rachmilewitz, E.A. Beta-Thalassaemia and sickle cell anaemia as paradigms of hypercoagulability. Br. J. Haematol. 2007, 139, 3–13. [Google Scholar] [CrossRef]
- Setty, B.N.Y.; Rao, A.K.; Stuart, M.J. Thrombophilia in sickle cell disease: The red cell connection. Blood 2001, 98, 3228–3233. [Google Scholar] [CrossRef] [Green Version]
- Noubouossie, D.; Key, N.S.; Ataga, K.I. Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev. 2016, 30, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Sparkenbaugh, E.; Pawlinski, R. Prothrombotic aspects of sickle cell disease. J. Thromb. Haemost. 2017, 15, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Faes, C.; Sparkenbaugh, E.M.; Pawlinski, R. Hypercoagulable state in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 301–318. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Malik, P. Role of the coagulation system in the pathogenesis of sickle cell disease. Blood Adv. 2019, 3, 3170–3180. [Google Scholar] [CrossRef]
- Toledo, S.L.d.O.; Guedes, J.V.M.; Alpoim, P.N.; Rios, D.R.A.; Pinheiro, M.d.B. Sickle cell disease: Hemostatic and inflammatory changes, and their interrelation. Clin. Chim. Acta 2019, 493, 129–137. [Google Scholar] [CrossRef]
- Conran, N.; De Paula, E.V. Thromboinflammatory mechanisms in sickle cell disease—challenging the hemostatic balance. Haematologica 2020, 105. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chen, C.; Brzoska, T.; Nguyen, J.; Wang, S.; Vercellotti, G.M.; Key, N.S.; Sundd, P.; Belcher, J.D.; Pawlinski, R. Thrombin activation of PAR-1 contributes to microvascular stasis in mouse models of sickle cell disease. Blood 2020, 135, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Kato, G.J. Hemolysis-associated hypercoagulability in sickle cell disease: The plot (and blood) thickens! Haematologica 2008, 93, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, M.L.N.; Murphy, A.V.; McMorris, S.; Jewell, F.G. Coagulation studies in haemolytic uraemic syndrome. Arch. Dis. Child. 1972, 47, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef]
- Kokoris, S.I.; Gavriilaki, E.; Miari, A.; Travlou, A.; Kyriakou, E.; Anagnostopoulos, A.; Grouzi, E. Renal involvement in paroxysmal nocturnal hemoglobinuria: An update on clinical features, pathophysiology and treatment. Hematology 2018, 23, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, R.; Ahluwalia, J.; Malhotra, P.; Mahapatra, M.; Varma, N.; Varma, S. Markers of thrombin generation and inflammation in patients with paroxysmal nocturnal hemoglobinuria. Indian J. Hematol. Blood Transfus. 2019. [Google Scholar] [CrossRef]
- Ozment, C.P.; Mamo, L.B.; Campbell, M.L.; Lokhnygina, Y.; Ghio, A.J.; Turi, J.L. Transfusion-related biologic effects and free hemoglobin, heme, and iron. Transfusion 2013, 53, 732–740. [Google Scholar] [CrossRef]
- Stolla, M.; Henrichs, K.; Cholette, J.M.; Pietropaoli, A.; Phipps, R.P.; Spinelli, S.; Blumberg, N. Increased free heme levels and red blood cell transfusions are associated with thrombosis in neonates and infants undergoing cardiac surgery for congenital heart disease. Blood 2014, 124, 2890. [Google Scholar] [CrossRef]
- Aleshnick, M.; Foley, J.H.; Keating, F.K.; Butenas, S. Procoagulant activity in stored units of red blood cells. Biochem. Biophys. Res. Commun. 2016, 474, 680–685. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Weisel, J.W. Role of red blood cells in haemostasis and thrombosis. ISBT Sci. Ser. 2017, 12, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Wagener, B.M.; Hu, P.J.; Oh, J.-Y.; Evans, C.A.; Richter, J.R.; Honavar, J.; Brandon, A.P.; Creighton, J.; Stephens, S.W.; Morgan, C.; et al. Role of heme in lung bacterial infection after trauma hemorrhage and stored red blood cell transfusion: A preclinical experimental study. PLoS Med. 2018, 15, e1002522. [Google Scholar] [CrossRef] [Green Version]
- Panch, S.R.; Montemayor-Garcia, C.; Klein, H.G. Hemolytic Transfusion Reactions. N. Engl. J. Med. 2019, 381, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Pietropaoli, A.P.; Henrichs, K.F.; Cholette, J.M.; Spinelli, S.L.; Phipps, R.P.; Refaai, M.A.; Blumberg, N. Total plasma heme concentration increases after red blood cell transfusion and predicts mortality in critically ill medical patients. Transfusion 2019, 59, 2007–2015. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Prudent, M.; D’alessandro, A. Red blood cell storage lesion: Causes and potential clinical consequences. Blood Transfus. 2019, 17, 27–52. [Google Scholar] [PubMed]
- Wagner, K.R.; Sharp, F.R.; Ardizzone, T.D.; Lu, A.; Clark, J.F. Heme and iron metabolism: Role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 2003, 23, 629–652. [Google Scholar] [CrossRef] [Green Version]
- Lok, J.; Leung, W.; Murphy, S.; Butler, W.; Noviski, N.; Lo, E.H. Intracranial hemorrhage: Mechanisms of secondary brain injury. Acta Neurochir. Suppl. 2011, 111, 63–69. [Google Scholar]
- Babu, R.; Bagley, J.H.; Di, C.; Friedman, A.H.; Adamson, C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage–induced secondary brain injury and as potential targets for intervention. Neurosurg. Focus 2012, 32, E8. [Google Scholar] [CrossRef] [Green Version]
- Michel, J.-B.; Delbosc, S.; Ho-Tin-Noé, B.; Leseche, G.; Nicoletti, A.; Meilhac, O.; Martin-Ventura, J.L. From intraplaque haemorrhages to plaque vulnerability. J. Cardiovasc. Med. 2012, 13, 628–634. [Google Scholar] [CrossRef]
- Robinson, S.R.; Dang, T.N.; Dringen, R.; Bishop, G.M. Hemin toxicity: A preventable source of brain damage following hemorrhagic stroke. Redox Rep. 2009, 14, 228–235. [Google Scholar] [CrossRef]
- Hu, S.; Hua, Y.; Keep, R.F.; Feng, H.; Xi, G. Deferoxamine therapy reduces brain hemin accumulation after intracerebral hemorrhage in piglets. Exp. Neurol. 2019, 318, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Stolla, M.; Henrichs, K.; Cholette, J.M.; Pietropaoli, A.; Phipps, R.P.; Spinelli, S.; Blumberg, N. Haem is associated with thrombosis in neonates and infants undergoing cardiac surgery for congenital heart disease. Vox Sang. 2018, 113, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Cholette, J.M.; Pietropaoli, A.P.; Henrichs, K.F.; Alfieris, G.M.; Powers, K.S.; Gensini, F.; Rubenstein, J.S.; Sweeney, D.; Phipps, R.; Spinelli, S.L.; et al. Elevated free hemoglobin and decreased haptoglobin levels are associated with adverse clinical outcomes, unfavorable physiologic measures, and altered inflammatory markers in pediatric cardiac surgery patients. Transfusion 2018, 58, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Egeberg, O. Blood coagulation and intravascular hemolysis. Scand. J. Clin. Lab. Investig. 1962, 14, 217–222. [Google Scholar] [CrossRef]
- Mannucci, P.M.; Lobina, G.F.; Caocci, L.; Dioguardi, N. Effect on blood coagulation of massive intravascular haemolysis. Blood 1969, 33, 207–213. [Google Scholar] [CrossRef]
- Vinchi, F.; Muckenthaler, M.U.; Da Silva, M.C.; Balla, G.; Balla, J.; Jeney, V. Atherogenesis and iron: From epidemiology to cellular level. Front. Pharmacol. 2014, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Cappellini, M.D. Coagulation in the pathophysiology of hemolytic anemias. Hematol. Am. Soc. Hematol. Educ. Progr. 2007, 2007, 74–78. [Google Scholar] [CrossRef] [Green Version]
- Riedler, G.; Straub, P.W.; Frick, P.G. The effects of acut intravascular hemolysis on coagulation and fibrinolysis. II. March hemoglobinemia and hemoglobinuria. Helv. Med. Acta 1968, 34, 217–222. [Google Scholar]
- Ataga, K.I. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica 2009, 94, 1481–1484. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.J.; Taylor, J.G. Pleiotropic effects of intravascular haemolysis on vascular homeostasis. Br. J. Haematol. 2010, 148, 690–701. [Google Scholar] [CrossRef] [Green Version]
- L’Acqua, C.; Hod, E. New perspectives on the thrombotic complications of haemolysis. Br. J. Haematol. 2015, 168, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Thein, S.L. Platelets at the crossroads of thrombosis, inflammation and haemolysis. Br. J. Haematol. 2018, 180, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Effenberger-Neidnicht, K.; Bornmann, S.; Jägers, J.; Patyk, V.; Kirsch, M. Microvascular stasis and hemolysis: Coincidence or causality? J. Inflamm. Res. 2019, 12, 109–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvagno, G.L.; Demonte, D.; Gelati, M.; Favaloro, E.J.; Lippi, G. Hemolysis induces a prothrombotic state in human plasma. In Proceedings of the International Society on Thrombosis and Haemostasis, Melbourne, Australia, 6–10 July 2019. [Google Scholar]
- Jeney, V.; Balla, G.; Balla, J. Red blood cell, hemoglobin and heme in the progression of atherosclerosis. Front. Physiol. 2014, 5, 379. [Google Scholar] [CrossRef] [Green Version]
- Brunson, A.; Lei, A.; Rosenberg, A.S.; White, R.H.; Keegan, T.; Wun, T. Increased incidence of VTE in sickle cell disease patients: Risk factors, recurrence and impact on mortality. Br. J. Haematol. 2017, 178, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, R.P.; Streiff, M.B.; Haywood, C.; Segal, J.B.; Lanzkron, S. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J. Thromb. Haemost. 2014, 12, 2010–2016. [Google Scholar] [CrossRef] [Green Version]
- Chantrathammachart, P.; Mackman, N.; Sparkenbaugh, E.; Wang, J.-G.; Parise, L.V.; Kirchhofer, D.; Key, N.S.; Pawlinski, R. Tissue factor promotes activation of coagulation and inflammation in a mouse model of sickle cell disease. Blood 2012, 120, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Setty, Y.; Key, N.S.; Rao, A.K.; Krishnan, S.; Gayen-Betal, S.; Stuart, M.J. Tissue factor procoagulant activity and coagulation activation in sickle cell disease: Relationship with biomarkers of hemolysis and inflammation. Blood 2010, 116, 4210. [Google Scholar] [CrossRef]
- Chekkal, M.; Rahal, M.C.A.; Moulasserdoun, K.; Seghier, F. Increased level of factor VIII and physiological inhibitors of coagulation in patients with sickle cell disease. Indian J. Hematol. Blood Transfus. 2017, 33, 235–238. [Google Scholar] [CrossRef]
- Abildgaard, C.F.; Simone, J.V.; Schulman, I. Factor-VIII (Antihaemophilic Factor) activity in sickle-cell anaemia. Br. J. Haematol. 1967, 13, 19–27. [Google Scholar] [CrossRef]
- Gordon, E.M.; Klein, B.L.; Berman, B.W.; Strandjord, S.E.; Simon, J.E.; Coccia, P.F. Reduction of contact factors in sickle cell disease. J. Pediatr. 1985, 106, 427–430. [Google Scholar] [CrossRef]
- Miller, R.L.; Verma, P.S.; Adams, R.G. Studies of the kallikrein-kinin system in patients with sickle cell anemia. J. Natl. Med. Assoc. 1983, 75, 551–556. [Google Scholar] [PubMed]
- Verma, P.S.; Adams, R.G.; Miller, R.L. Reduced plasma kininogen concentration during sickle cell crisis. Res. Commun. Chem. Pathol. Pharmacol. 1983, 41, 313–322. [Google Scholar] [PubMed]
- Nsiri, B.; Gritli, N.; Bayoudh, F.; Messaoud, T.; Fattoum, S.; Machghoul, S. Abnormalities of coagulation and fibrinolysis in homozygous sickle cell disease. Hematol. Cell Ther. 1996, 38, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Tomer, A.; Harker, L.A.; Kasey, S.; Eckman, J.R. Thrombogenesis in sickle cell disease. J. Lab. Clin. Med. 2001, 137, 398–407. [Google Scholar] [CrossRef]
- Westerman, M.P.; Green, D.; Gilman-Sachs, A.; Beaman, K.; Freels, S.; Boggio, L.; Allen, S.; Zuckerman, L.; Schlegel, R.; Williamson, P. Antiphospholipid antibodies, proteins C and S, and coagulation changes in sickle cell disease. J. Lab. Clin. Med. 1999, 134, 352–362. [Google Scholar] [CrossRef]
- Leichtman, D.A.; Brewer, G.J. Elevated plasma levels of fibrinopeptide a during sickle cell anemia pain crisis–evidence for intravascular coagulation. Am. J. Hematol. 1978, 5, 183–190. [Google Scholar] [CrossRef]
- Slater, S.D.; Prentice, C.R.M.; Bain, W.H.; Briggs, J.D. Fibrinogen-fibrin degradation product levels in different types of intravascular haemolysis. Br. Med. J. 1973, 3, 471–473. [Google Scholar] [CrossRef] [Green Version]
- Sack, E.S.; Nefa, O.M. Fibrinogen and fibrin degradation products in hemolytic transfusion reactions. Transfusion 1970, 10, 317–321. [Google Scholar] [CrossRef]
- Shiu, Y.-T.; Udden, M.M.; McIntire, L.V. Perfusion with sickle erythrocytes up-regulates ICAM-1 andVCAM-1 gene expression in cultured human endothelial cells. Blood 2000, 95, 3232–3241. [Google Scholar] [CrossRef]
- Conran, N.; Fattori, A.; Saad, S.T.O.; Costa, F.F. Increased levels of soluble ICAM-1 in the plasma of sickle cell patients are reversed by hydroxyurea. Am. J. Hematol. 2004, 76, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Kucukal, E.; Ilich, A.; Key, N.S.; Little, J.A.; Gurkan, U.A. Red blood cell adhesion to heme-activated endothelial cells reflects clinical phenotype in sickle cell disease. Am. J. Hematol. 2018, 93, 1050–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hobbs, W.E.; Le, J.; Lenting, P.J.; de Groot, P.G.; López, J.A. The rate of hemolysis in sickle cell disease correlates with the quantity of active von Willebrand factor in the plasma. Blood 2011, 117, 3680–3683. [Google Scholar] [CrossRef] [PubMed]
- Voskaridou, E.; Larrisi, K.; Politou, M.; Komninaka, V.; Apostolakou, F.; Skevaki, C.; Giannaki, M.; Papassotiriou, I.; Terpos, E. Increased von Willebrand factor and high circulating placental growth factor correlate with inflammation and iron overload in patients with compound heterozygous sickle cell and beta-Thalassemia. Blood 2014, 124, 1392. [Google Scholar] [CrossRef]
- Voskaridou, E.; Aimilia, M.; Flevari, P.; Dimopoulou, M.; Komninaka, V.; Repa, K.; Papassotiriou, I. Soluble P-selectin levels in patients with sickle cell disease reflect platelets’ activation rather than endothelial dysfunction. Blood 2019, 134, 4829. [Google Scholar] [CrossRef]
- Guo, Y.; Uy, T.; Wandersee, N.; Scott, J.P.; Weiler, H.; Holzhauer, S.; Retherford, D.; Foster, T.; Hillery, C. The protein C pathway in human and murine sickle cell disease: Alterations in protein C, thrombomodulin (TM), and endothelial protein C receptor (EPCR) at baseline and during acute vaso-occlusion. Blood 2008, 112, 538. [Google Scholar] [CrossRef]
- Ruiz, M.A.; Shah, B.N.; Han, J.; Raslan, R.; Gordeuk, V.R.; Saraf, S.L. Thrombomodulin and endothelial dysfunction in sickle cell anemia. Blood 2019, 134, 3558. [Google Scholar] [CrossRef]
- Bayazit, A.K.; Kilinç, Y. Natural coagulation inhibitors (Protein C, protein S, antithrombin) in patients with sickle cell anemia in a steady state. Pediatr. Int. 2001, 43, 592–596. [Google Scholar] [CrossRef]
- Schnog, J.B.; Mac Gillavry, M.R.; Van Zanten, A.P.; Meijers, J.C.M.; Rojer, R.A.; Duits, A.J.; Cate, H.; Brandjes, D.P.M. Protein C and S and inflammation in sickle cell disease. Am. J. Hematol. 2004, 76, 26–32. [Google Scholar] [CrossRef]
- El-Hazmi, M.A.F.; Warsy, A.S.; Bahakim, H. Blood proteins C and S in sickle cell disease. Acta Haematol. 1993, 90, 114–119. [Google Scholar] [CrossRef]
- Khanduri, U.; Gravell, D.; Christie, B.S.; Al Lamki, Z.; Zachariah, M.; Cherian, E. Reduced protein C levels—A contributory factor for stroke in sickle cell disease. Thromb. Haemost. 1998, 79, 879–880. [Google Scholar] [PubMed]
- Van der Merwe, L.; Reyers, F. The effect of hemolysis on plasma antithrombin activity as determined by a chromogenic method. Vet. Clin. Pathol. 2007, 36, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Helms, C.C.; Marvel, M.; Zhao, W.; Stahle, M.; Vest, R.; Kato, G.J.; Lee, J.S.; Christ, G.; Gladwin, M.T.; Hantgan, R.R.; et al. Mechanisms of hemolysis-associated platelet activation. J. Thromb. Haemost. 2013, 11, 2148–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villagra, J.; Shiva, S.; Hunter, L.A.; Machado, R.F.; Gladwin, M.T.; Kato, G.J. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 2007, 110, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Deng, X.; Ma, R.; Dong, Z.; Wang, F.; Shi, J. The exposure of phosphatidylserine influences procoagulant activity in retinal vein occlusion by microparticles, blood cells, and endothelium. Oxid. Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Zwaal, R.F.A.; Schroit, A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef]
- Mense, S.M.; Zhang, L.L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L. (Ed.) Heme Biology: The Secret Life of Heme in Regulating Diverse Biological Processes; World Scientific: Singapore, 2011. [Google Scholar]
- Kühl, T.; Imhof, D. Regulatory Fe II/III Heme: The reconstruction of a molecule’s biography. ChemBioChem 2014, 15, 2024–2035. [Google Scholar] [CrossRef]
- Shimizu, T.; Lengalova, A.; Martinek, V.; Martinkova, M. Heme: Emergent roles of heme in signal transduction, functional regulation and as catalytic centres. Chem. Soc. Rev. 2019, 48, 5624–5657. [Google Scholar] [CrossRef]
- Korolnek, T.; Hamza, I. Like iron in the blood of the people: The requirement for heme trafficking in iron metabolism. Front. Pharmacol. 2014, 5, 126. [Google Scholar] [CrossRef] [Green Version]
- Sawicki, K.T.; Chang, H.; Ardehali, H. Role of heme in cardiovascular physiology and disease. J. Am. Heart Assoc. 2015, 4, e001138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandro, A.; Zolla, L. Proteomic analysis of red blood cells and the potential for the clinic: What have we learned so far? Expert Rev. Proteom. 2017, 14, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; Bocedi, A.; Visca, P.; Altruda, F.; Tolosano, E.; Beringhelli, T.; Fasano, M. Hemoglobin and heme scavenging. IUBMB Life 2005, 57, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Rayes, J.; Lacroix-Desmazes, S.; Dimitrov, J.D. Heme: Modulator of plasma systems in hemolytic diseases. Trends Mol. Med. 2016, 22, 200–213. [Google Scholar] [CrossRef]
- Frimat, M.; Boudhabhay, I.; Roumenina, L.T. Hemolysis derived products toxicity and endothelium: Model of the second hit. Toxins 2019, 11, 660. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Martins, R.; Knapp, S. Heme and hemolysis in innate immunity: Adding insult to injury. Curr. Opin. Immunol. 2018, 50, 14–20. [Google Scholar] [CrossRef]
- Rapido, F. The potential adverse effects of haemolysis. Blood Transfus. 2017, 15, 218–221. [Google Scholar]
- Donegan, R.K.; Moore, C.M.; Hanna, D.A.; Reddi, A.R. Handling heme: The mechanisms underlying the movement of heme within and between cells. Free Radic. Biol. Med. 2019, 133, 88–100. [Google Scholar] [CrossRef]
- Cooper, C.E.; Schaer, D.J.; Buehler, P.W.; Wilson, M.T.; Reeder, B.J.; Silkstone, G.; Svistunenko, D.A.; Bulow, L.; Alayash, A.I. Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine β145. Antioxid. Redox Signal. 2013, 18, 2264–2273. [Google Scholar] [CrossRef]
- Schaer, D.J.; Vinchi, F.; Ingoglia, G.; Tolosano, E.; Buehler, P.W. Haptoglobin, hemopexin, and related defense pathways—basic science, clinical perspectives, and drug development. Front. Physiol. 2014, 5, 415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rother, R.P.; Bell, L.; Hillmen, P.; Gladwin, M.T. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. JAMA 2005, 293, 1653. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; De Simone, G.; Ciaccio, C.; D’Orso, S.; Coletta, M.; Ascenzi, P. Haptoglobin: From hemoglobin scavenging to human health. Mol. Aspects Med. 2020, 73, 100851. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid. Med. Cell. Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruitier, I.; Garreau, I.; Lacroix, A.; Cupo, A.; Piot, J.M. Proteolytic degradation of hemoglobin by endogenous lysosomal proteases gives rise to bioactive peptides: Hemorphins. FEBS Lett. 1999, 447, 81–86. [Google Scholar] [CrossRef] [Green Version]
- Schaer, C.A.; Schoedon, G.; Imhof, A.; Kurrer, M.O.; Schaer, D.J. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ. Res. 2006, 99, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and Hemopexin in Heme Detoxification and Iron Recycling. In Acute Phase Proteins—Regulation and Functions of Acute Phase Proteins; Veas, F., Ed.; InTech: London, UK, 2011; pp. 261–288. [Google Scholar]
- Vijayan, V.; Wagener, F.A.D.T.G.; Immenschuh, S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem. Pharmacol. 2018, 153, 159–167. [Google Scholar] [CrossRef]
- Duvigneau, J.C.; Esterbauer, H.; Kozlov, A.V. Role of heme oxygenase as a modulator of heme-mediated Pathways. Antioxidants 2019, 8, 475. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [Green Version]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umbreit, J. Methemoglobin—It’s not just blue: A concise review. Am. J. Hematol. 2007, 82, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, Z.; Carlos, A.R.; Yuan, X.; Aires-da-Silva, F.; Stocker, R.; Maghzal, G.J.; Leal, S.S.; Gomes, C.M.; Todorovic, S.; Iranzo, O.; et al. Characterization of plasma labile heme in hemolytic conditions. FEBS J. 2017, 284, 3278–3301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassa, T.; Jana, S.; Meng, F.; Alayash, A.I. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. FEBS Open Bio 2016, 6, 876–884. [Google Scholar] [CrossRef]
- Schaer, D.J.; Buehler, P.W.; Alayash, A.I.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef] [Green Version]
- Ascenzi, P.; di Masi, A.; Fanali, G.; Fasano, M. Heme-based catalytic properties of human serum albumin. Cell Death Discov. 2015, 1, 15025. [Google Scholar] [CrossRef] [Green Version]
- Kamal, J.K.A.; Behere, D.V. Binding of heme to human serum albumin: Steady-state fluorescence, circular dichroism and optical difference spectroscopic studies. Indian J. Biochem. Biophys. 2005, 42, 7–12. [Google Scholar]
- Morgan, W.T.; Heng Liem, H.; Sutor, R.P.; Muller-Eberhard, U. Transfer of heme from heme-albumin to hemopexin. Biochim. Biophys. Acta Gen. Subj. 1976, 444, 435–445. [Google Scholar] [CrossRef]
- Hrkal, Z.; Vodrazka, Z.; Kalousek, I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur. J. Biochem. 1974, 43, 73–78. [Google Scholar] [CrossRef]
- Hvidberg, V.; Maniecki, M.B.; Jacobsen, C.; Højrup, P.; Møller, H.J.; Moestrup, S.K. Identification of the receptor scavenging hemopexin-heme complexes. Blood 2005, 106, 2572–2579. [Google Scholar] [CrossRef]
- Leff, J.A.; Kennedy, D.A.; Terada, L.S.; Emmett, M.; McCutchan, H.J.; Walden, D.L.; Repine, J.E. Reperfusion of ischemic skeletal muscle causes erythrocyte hemolysis and decreases subsequent oxidant-mediated lung injury. J. Lab. Clin. Med. 1991, 118, 352–358. [Google Scholar] [PubMed]
- Strobel, E. Hemolytic transfusion reactions. Transfus. Med. Hemother. 2008, 35, 346–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presley, T.; Bain, L.; Ballas, S.; Nichols, J.; Sabio, H.; Gladwin, M.; Kato, G.J.; Kim-Shapiro, D. The mechanism of hemolysis in sickle cell anemia. Blood 2008, 112, 1439. [Google Scholar] [CrossRef]
- Wu, B.; Wu, Y.; Tang, W. Heme Catabolic Pathway in Inflammation and Immune Disorders. Front. Pharmacol. 2019, 10, 825. [Google Scholar] [CrossRef] [Green Version]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, B.N.; Alves, L.S.; Fernández, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graça-Souza, A.V.; Bozza, M.T. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 2007, 282, 24430–24436. [Google Scholar] [CrossRef] [Green Version]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, B.; Hauser, C.J.; Otterbein, L.E. Heme as a danger molecule in pathogen recognition. Free Radic. Biol. Med. 2015, 89, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Hod, E.A. Consequences of hemolysis: Pro-inflammatory cytokine response to erythrophagocytosis. Transfus. Clin. Biol. 2019, 26, 125–127. [Google Scholar] [CrossRef]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014, 111, E4110–E4118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nader, E.; Romana, M.; Connes, P. The red blood cell—Inflammation vicious circle in sickle cell disease. Front. Immunol. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humayun, F.; Domingo-Fernández, D.; Paul George, A.A.; Hopp, M.-T.; Syllwasschy, B.F.; Detzel, M.S.; Hoyt, C.T.; Hofmann-Apitius, M.; Imhof, D. A computational approach for mapping heme biology in the context of hemolytic disorders. Front. Bioeng. Biotechnol. 2020, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, W.H. The relation of hematin to pathological pigment formation. J. Exp. Med. 1911, 14, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Küster, W. Beiträge zur Kenntnis des Bilirubins und Hämins. Hoppe Seylers Z. Physiol. Chem. 1912, 82, 463. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.H.; Loevenhart, A.S. The effect of hemati on the circulation and respiration. J. Exp. Med. 1913, 18, 107–112. [Google Scholar] [CrossRef]
- Brown, W.H. The renal complications of hematin intoxification and their relation to Malaria. Arch. Intern. Med. 1913, 12, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Brown, W.H. Malarial pigment (hematin) as an active factor in the production of the blood picture of malaria. J. Exp. Med. 1913, 18, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W.A.D.; Morrison, D.B.; Williams, E.F. Pathologic changes following injections of ferrihemate (hematin) in dogs. Arch. Pathol. 1942, 33, 589–602. [Google Scholar]
- Corcoran, A.C.; Page, I.H. Renal damage from ferroheme pigments myoglobin, hemoglobin, hematin. Tex. Rep. Biol. Med. 1945, 3, 528–544. [Google Scholar]
- Gessler, U.; Loreth, A.; Schröder, K.; Steinhausen, M. Experimentelle Untersuchungen über die glomeruläre Filtration anurischer Ratten nach Hämatinvergiftung. Klin. Wochenschr. 1966, 44, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Bonkowsky, H.L.; Tschudy, D.P.; Collins, A.; Doherty, J.; Bossenmaier, I.; Cardinal, R.; Watson, C.J. Repression of the overproduction of porphyrin precursors in acute intermittent porphyria by intravenous infusions of hematin. Proc. Natl. Acad. Sci. USA 1971, 68, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lips, D.L.; Pierach, C.A.; Edwards, P.S. Hematin toxicity in rats. Toxicol. Lett. 1978, 2, 329–332. [Google Scholar] [CrossRef]
- Watson, C.J.; Pierach, C.A.; Bossenmaier, I.; Cardinal, R. Postulated deficiency of hepatic heme and repair by hematin infusions in the “inducible” hepatic porphyrias. Proc. Natl. Acad. Sci. USA 1977, 74, 2118–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, C.J.; Pierach, C.A.; Bossenmaier, I.; Cardinal, R. The effect of hematin in “inducible” hepatic porphyria. In Diagnosis and Therapy of Porphyrias and Lead Intoxication; Springer: Berlin, Germany, 1978; pp. 100–102. [Google Scholar]
- Anderson, K.E.; Collins, S. Open-label study of hemin for acute porphyria: Clinical practice implications. Am. J. Med. 2006, 119, e1–e6. [Google Scholar] [CrossRef]
- Dhar, G.J.; Bossenmaier, I.; Petryka, Z.J.; Cardinal, R.; Watson, C.J. Effect of hematin in hepatic porphyria: Further studies. Ann. Intern. Med. 1975, 83, 20–30. [Google Scholar] [CrossRef]
- Lamon, J.M.; Frykholm, B.C.; Hess, R.A.; Tschudy, D.P. Hematin therapy for acute porphyria. Medicine 1979, 58, 252–269. [Google Scholar] [CrossRef]
- Morris, D.L.; Dudley, M.D.; Pearson, R.D. Coagulopathy associated with hematin treatment for acute intermittent porphyria. Ann. Intern. Med. 1981, 95, 700–701. [Google Scholar] [CrossRef]
- McColl, K.E.; Moore, M.R.; Thompson, G.G.; Goldberg, A. Treatment with haematin in acute hepatic porphyria. Q. J. Med. 1981, 50, 161–174. [Google Scholar]
- Bloomer, J.R.; Pierach, C.A. Effect of hematin administration to patients with protoporphyria and liver disease. Hepatology 1982, 2, 817–821. [Google Scholar] [CrossRef]
- Simionatto, C.S.; Cabal, R.; Jones, R.L.; Galbraith, R.A. Thrombophlebitis and disturbed hemostasis following administration of intravenous hematin in normal volunteers. Am. J. Med. 1988, 85, 538–540. [Google Scholar] [CrossRef]
- Goetsch, C.A.; Bissell, D.M. Instability of hematin used in the treatment of acute hepatic porphyria. N. Engl. J. Med. 1986, 315, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Tokola, O.; Lindén, I.-B. Haem arginate: A new stable haem compound. J. Pharm. Pharmacol. 1987, 39, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Balla, G.; Kakuk, G.; Nath, K.A.; Jacob, H.S.; Vercellotti, G.M. Heme arginate and the endothelium: Mechanism for its safety in porphyria. J. Investig. Med. 1996, 44, 214a. [Google Scholar]
- Balla, J.; Balla, G.; Jeney, V.; Kakuk, G.; Jacob, H.S.; Vercellotti, G.M. Ferriporphyrins and endothelium: A 2-edged sword—promotion of oxidation and induction of cytoprotectants. Blood 2000, 95, 3442–3450. [Google Scholar] [CrossRef]
- Mustajoki, P.; Tenhunen, R.; Tokola, O.; Gothoni, G. Haem arginate in the treatment of acute hepatic porphyrias. Br. Med. J. Clin. Res. Ed. 1986, 293, 538–539. [Google Scholar] [CrossRef] [Green Version]
- Ruutu, T.; Volin, L.; Tenhunen, R. Haem arginate as a treatment for myelodysplastic syndromes. Br. J. Haematol. 1987, 65, 425–428. [Google Scholar] [CrossRef]
- Marsden, J.T.; Guppy, S.; Stein, P.; Cox, T.M.; Badminton, M.; Gardiner, T.; Barth, J.H.; Stewart, M.F.; Rees, D.C. Audit of the use of regular haem arginate infusions in patients with acute porphyria to prevent recurrent symptoms. JIMD Rep. 2015, 22, 57–65. [Google Scholar]
- Mustajoki, P.; Nordmann, Y. Early administration of heme arginate for acute porphyric attacks. Arch. Intern Med. 1993, 153, 2004–2008. [Google Scholar] [CrossRef]
- Herrick, A.L.; Moore, M.R.; Mccoll, K.E.L.; Cook, A.; Goldberg, A. Controlled trial of haem arginate in acute hepatic porphyria. Lancet 1989, 333, 1295–1297. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Healey, J.F.; Lourie, A.N.; Gerron, G.G. Intravenous heme-albumin in acute intermittent porphyria: Evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am. J. Gastroenterol. 1991, 86, 1050–1056. [Google Scholar] [PubMed]
- Anderson, K.E.; Bonkovsky, H.L.; Bloomer, J.R.; Shedlofsky, S.I. Reconstitution of hematin for intravenous infusion. Ann. Intern. Med. 2006, 144, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajra, A.; Vajpayee, N.; Singh, M.; Coyle, T.E.; Wright, J. Hematin induced coagulopathy in acute intermittent porphyria: A case report. Blood 2000, 96, 82b. [Google Scholar]
- Glueck, R.; Green, D.; Cohen, I.; Ts’ao, C. Hematin: Unique effects on hemostasis. Blood 1983, 61, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshal, M.; Reyes Gil, M. Activated partial thromboplastin time. In Transfusion Medicine and Hemostasis: Clinical and Laboratory Aspects; Shaz, B.H., Hillyer, C.D., Gil, M.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 779–781. [Google Scholar]
- Zimring, J.C. Prothrombin time. In Transfusion Medicine and Hemostasis: Clinical and Laboratory Aspects; Hillyer, C.D., Shaz, B.H., Zimring, J.C., Abshire, T.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 607–610. [Google Scholar]
- Yuan, S.; Ferrell, C.; Chandler, W.L. Comparing the prothrombin time INR versus the APTT to evaluate the coagulopathy of acute trauma. Thromb. Res. 2007, 120, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.D. The effect of saccharin ingestion on blood coagulation and the in vitro anticoagulant effect of saccharin and of ferriheme. J. Am. Pharm. Assoc. Sci. Ed. 1947, 36, 225–228. [Google Scholar] [CrossRef]
- Green, D.; Reynolds, N.; Klein, J.; Kohl, H.; Ts’ao, C.H. The inactivation of hemostatic factors by hematin. J. Lab. Clin. Med. 1983, 102, 361–369. [Google Scholar] [PubMed]
- Jones, R.L. Hematin-derived anticoagulant. Generation in vitro and in vivo. J. Exp. Med. 1986, 163, 724–739. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.G.; Wagner, M.; Kaplan, A.P.; Silverberg, M.; Grady, R.W.; Liem, H.; Muller-Eberhard, U. Activation of factor XII-dependent pathways in human plasma by hematin and protoporphyrin. J. Clin. Investig. 1985, 76, 413–419. [Google Scholar] [CrossRef] [Green Version]
- Volin, L.; Rasi, V.; Vahtera, E.; Tenhunen, R. Heme arginate: Effects on hemostasis. Blood 1988, 71, 625–628. [Google Scholar] [CrossRef] [Green Version]
- Green, D.; Ts’ao, C. Hematin: Effects on hemostasis. J. Lab. Clin. Med. 1990, 115, 144–147. [Google Scholar] [PubMed]
- Huang, Y.; Komatsu, T.; Nakagawa, A.; Tsuchida, E.; Kobayashi, S. Compatibility in vitro of albumin-heme (O2 carrier) with blood cell components. J. Biomed. Mater. Res. 2003, 66, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Rochefort, G.Y.; Libgot, R.; Desbuards, N.; Schlecht, D.; Halimi, J.M.; Ossant, F.; Eder, V.; Antier, D. Effect of the heme oxygenase inducer hemin on blood haemostasis measured by high-frequency ultrasound. Clin. Exp. Pharmacol. Physiol. 2007, 34, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Desbuards, N.; Rochefort, G.Y.; Schlecht, D.; Machet, M.-C.; Halimi, J.-M.; Eder, V.; Hyvelin, J.-M.; Antier, D. Heme oxygenase-1 inducer hemin prevents vascular thrombosis. Thromb. Haemost. 2007, 98, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Marcantoni, E.; Di Francesco, L.; Dovizio, M.; Bruno, A.; Patrignani, P. Novel Insights into the vasoprotective role of heme oxygenase-1. Int. J. Hypertens. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Pae, H.-O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.-H.; Choi, Y.K.; Lee, B.-S.; Kim, S.R.; Chung, H.-T. Heme oxygenase in the regulation of vascular biology: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 14, 137–167. [Google Scholar] [CrossRef] [Green Version]
- Fei, D.; Meng, X.; Zhao, M.M.; Kang, K.A.I.; Tan, G.; Pan, S.; Luo, Y.; Liu, W.E.N.; Nan, C.; Jiang, H.; et al. Enhanced induction of heme oxygenase-1 suppresses thrombus formation and affects the protein C system in sepsis. Transl. Res. 2012, 159, 99–109. [Google Scholar] [CrossRef]
- Hassaan, P.S.; Mehanna, R.A.; Dief, A.E. The potential role of hemopexin and heme oxygenase-1 inducer in a model of sepsis. Physiol. J. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- De Souza, G.R.; Hounkpe, B.W.; Fiusa, M.M.L.; Colella, M.P.; Annichino-Bizzacchi, J.M.; Traina, F.; Costa, F.F.; De Paula, E.V. Tissue factor-dependent coagulation activation by heme: A thromboelastometry study. PLoS ONE 2017, 12, 1–10. [Google Scholar] [CrossRef]
- Hopp, M.-T.; Alhanafi, N.; Paul George, A.A.; Hamedani, N.S.; Biswas, A.; Oldenburg, J.; Pötzsch, B.; Imhof, D. Molecular insights and functional consequences of the interaction of heme with activated protein C. Antioxid. Redox Signal. 2021, 34, 32–48. [Google Scholar] [CrossRef]
- Spronk, H.M.H.; Govers-Riemslag, J.W.P.; Ten Cate, H. The blood coagulation system as a molecular machine. BioEssays 2003, 25, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism action of platelets and crucial blood coagulation pathways in hemostasis. Int. J. Hematol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar]
- Peterson, D.; Gerrard, J.; Glover, S.; Rao, G.; White, J. Epinephrine reduction of heme: Implication for understanding the transmission of an agonist stimulus. Science 1982, 215, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.; Creter, D.; Cohen, A.; Djaldetti, M. Haemin affects platelet aggregation and lymphocyte mitogenicity in whole blood incubations. Cytobios 1983, 38, 33–38. [Google Scholar]
- Neely, S.M.; Gardner, D.; Reynolds, N.; Green, D.; Ts’ao, C. Mechanism and characteristics of platelet activation by haematin. Br. J. Haematol. 1984, 58, 305–316. [Google Scholar] [CrossRef]
- Volin, L.; Ruutu, T.; Knuutila, S.; Tenhunen, R. Heme arginate treatment for myelodysplastic syndromes. Leuk. Res. 1988, 12, 423–431. [Google Scholar] [CrossRef]
- Komatsu, T.; Huang, Y.; Wakamoto, S.; Abe, H.; Fujihara, M.; Azuma, H.; Ikeda, H.; Yamamoto, H.; Horinouchi, H.; Kobayashi, K.; et al. Influence of O2-carrying plasma hemoprotein “albumin-heme” on complement system and platelet activationin vitro and physiological responses to exchange transfusion. J. Biomed. Mater. Res. Part A 2007, 81, 821–826. [Google Scholar] [CrossRef]
- Peng, L.; Mundada, L.; Stomel, J.M.; Liu, J.J.; Sun, J.; Yet, S.-F.; Fay, W.P. Induction of heme oxygenase-1 expression inhibits platelet-dependent thrombosis. Antioxid. Redox Signal. 2004, 6, 729–735. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; SharathBabu, B.N.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S.; Mugesh, G. The role of reactive oxygen species and ferroptosis in heme-mediated activation of human platelets. ACS Chem. Biol. 2018, 13, 1996–2002. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Key, N.S. Microparticles in sickle cell anaemia: Promise and pitfalls. Br. J. Haematol. 2016, 174, 16–29. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Bourne, J.H.; Colicchia, M.; Di, Y.; Martin, E.; Slater, A.; Roumenina, L.T.; Dimitrov, J.D.; Watson, S.P.; Rayes, J. Heme induces human and mouse platelet activation through C-type-lectin-like receptor-2. Haematologica 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hao, H.; Leeper, N.J.; Zhu, L. Thrombotic regulation from the endothelial cell perspectives. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e90–e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neely, S.M.; Gardner, D.V.; Green, D.; Ts’ao, C.H. Effect of hematin on endothelial cells and endothelial cell-platelet interactions. Am. J. Pathol. 1984, 115, 390–396. [Google Scholar]
- Balla, G.; Vercollotti, G.; Muller-Eberhard, U.; Eaton, J.; Jacob, H.S. Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Investig. 1991, 64, 648–655. [Google Scholar]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Woollard, K.J.; Sturgeon, S.; Chin-Dusting, J.P.F.; Salem, H.H.; Jackson, S.P. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J. Biol. Chem. 2009, 284, 13110–13118. [Google Scholar] [CrossRef] [Green Version]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef] [Green Version]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M.; et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.-C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [Green Version]
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin causes lung microvascular endothelial barrier dysfunction by necroptotic cell death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314. [Google Scholar] [CrossRef] [PubMed]
- May, O.; Merle, N.S.; Grunenwald, A.; Gnemmi, V.; Leon, J.; Payet, C.; Robe-Rybkine, T.; Paule, R.; Delguste, F.; Satchell, S.C.; et al. Heme drives susceptibility of glomerular endothelium to complement overactivation due to inefficient upregulation of heme oxygenase-1. Front. Immunol. 2018, 9, 3008. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3, e96910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrnes, J.R.; Wolberg, A.S. Red blood cells in thrombosis. Blood 2017, 130, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.C.; Fitch, C.D. Mechanism of hemolysis induced by ferriprotoporphyrin IX. J. Clin. Investig. 1981, 68, 672–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaklai, N.; Shviro, Y.; Rabizadeh, E.; Kirschner-Zilber, I. Accumulation and drainage of hemin in the red cell membrane. Biochim. Biophys. Acta Biomembr. 1985, 821, 355–366. [Google Scholar] [CrossRef]
- Liu, S.C.; Zhai, S.; Lawler, J.; Palek, J. Hemin-mediated dissociation of erythrocyte membrane skeletal proteins. J. Biol. Chem. 1985, 260, 12234–12239. [Google Scholar] [CrossRef]
- Das, D.; Patra, M.; Chakrabarti, A. Binding of hemin, hematoporphyrin, and protoporphyrin with erythroid spectrin: Fluorescence and molecular docking studies. Eur. Biophys. J. 2015, 44, 171–182. [Google Scholar] [CrossRef]
- Elstad, M.R.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. The interaction of leukocytes with platelets in blood coagulation. Curr. Opin. Hematol. 1995, 2, 47–54. [Google Scholar] [CrossRef]
- Swystun, L.L.; Liaw, P.C. The role of leukocytes in thrombosis. Blood 2016, 128, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Miki, T.; Hashiguchi, N.; Tabe, Y.; Nagaoka, I. Is the neutrophil a ‘prima donna’ in the procoagulant process during sepsis? Crit. Care 2014, 18, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.J.; Winslow, R.M. Effects of extraerythrocytic hemoglobin and its components on mononuclear cell procoagulant activity. J. Lab. Clin. Med. 1992, 119, 176–182. [Google Scholar] [PubMed]
- Wagener, F.A.D.T.G.; Eggert, A.; Boerman, O.C.; Oyen, W.J.G.; Verhofstad, A.; Abraham, N.G.; Adema, G.; Van Kooyk, Y.; De Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar] [CrossRef]
- Arruda, M.A.; Rossi, A.G.; de Freitas, M.S.; Barja-Fidalgo, C.; Graça-Souza, A.V. Heme inhibits human neutrophil apoptosis: Involvement of phosphoinositide 3-kinase, MAPK, and NF-κB. J. Immunol. 2004, 173, 2023–2030. [Google Scholar] [CrossRef] [PubMed]
- Graça-Souza, A.V.; Arruda, M.A.B.; De Freitas, M.S.; Barja-Fidalgo, C.; Oliveira, P.L. Neutrophil activation by heme: Implications for inflammatory processes. Blood 2002, 99, 4160–4165. [Google Scholar] [CrossRef] [Green Version]
- Wagener, F.A.D.T.G.; van Beurden, H.E.; von den Hoff, J.W.; Adema, G.J.; Figdor, C.G. The heme-heme oxygenase system: A molecular switch in wound healing. Blood 2003, 102, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Ohbuchi, A.; Kono, M.; Kitagawa, K.; Takenokuchi, M.; Imoto, S.; Saigo, K. Quantitative analysis of hemin-induced neutrophil extracellular trap formation and effects of hydrogen peroxide on this phenomenon. Biochem. Biophys. Rep. 2017, 11, 147–153. [Google Scholar] [CrossRef]
- Belcher, J.D.; Nguyen, J.; Chen, C.; Smith, A.; Alayash, A.I.; Nair, S.L.; Somani, A.; Hebbel, R.P.; Vercellotti, G.M. Plasma hemoglobin and heme trigger Weibel Palade body exocytosis and vaso-occlusion in transgenic sickle mice. Blood 2011, 118, 896. [Google Scholar] [CrossRef]
- Howell, W.-H. Note relative a l’action photodynamique de l’hématoporphyrine sur le fibrinogéne. Arch. Int. Physiol. 1921, 18, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Boyd, M.J. Hematoporphyrin, an artificial proteolytic enzyme. J. Biol. Chem. 1933, 103, 249. [Google Scholar] [CrossRef]
- Zieve, P.; Solomon, H. Effect of hematoporphyrin and light on human fibrinogen. Am. J. Physiol. Content 1966, 210, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Musser, D.A.; Wagner, J.M.; Weber, F.J.; Datta-Gupta, N. The effect of tumor localizing porphyrins on the conversion of fibrinogen to fibrin. Res. Commun. Chem. Pathol. Pharmacol. 1979, 26, 357–382. [Google Scholar]
- Musser, D.A.; Wagner, J.M.; Weber, F.J.; Datta-Gupta, N. The binding of tumor localizing porphyrins to a fibrin matrix and their effects following photoirradiation. Res. Commun. Chem. Pathol. Pharmacol. 1980, 28, 505–525. [Google Scholar]
- Nielsen, V.G.; Cohen, J.B.; Malayaman, S.N.; Nowak, M.; Vosseller, K. Fibrinogen is a heme-associated, carbon monoxide sensing molecule: A preliminary report. Blood Coagul. Fibrinolysis 2011, 22, 443–447. [Google Scholar] [CrossRef]
- Barrera, V.; Skorokhod, O.A.; Baci, D.; Gremo, G.; Arese, P.; Schwarzer, E. Host fibrinogen stably bound to hemozoin rapidly activates monocytes via TLR-4 and CD11b/CD18-integrin: A new paradigm of hemozoin action. Blood 2011, 117, 5674–5682. [Google Scholar] [CrossRef] [Green Version]
- Orino, K. Functional binding analysis of human fibrinogen as an iron- and heme-binding protein. BioMetals 2013, 26, 789–794. [Google Scholar] [CrossRef]
- Ke, Z.; Huang, Q. Haem-assisted dityrosine-cross-linking of fibrinogen under non-thermal plasma exposure: One important mechanism of facilitated blood coagulation. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Hou, T.; Zhang, Y.; Wu, T.; Wang, M.; Zhang, Y.; Li, R.; Wang, L.; Xue, Q.; Wang, S. Label-free detection of fibrinogen based on fibrinogen-enhanced peroxidase activity of fibrinogen-hemin composite. Analyst 2018, 143, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.B.; Biggs, R.; Denson, K.W.E. “Inhibitor like” effects of hematoporphyrin on factor VIII (antihaemophilic globulin). Thromb. Haemost. 1967, 17, 299–306. [Google Scholar] [CrossRef]
- Green, D.; Furby, F.H.; Berndt, M.C. The interaction of the VIII/von Willebrand factor complex with hematin. Thromb. Haemost. 1986, 56, 277–282. [Google Scholar] [PubMed]
- Repessé, Y.; Dimitrov, J.D.; Peyron, I.; Moshai, E.F.; Kiger, L.; Dasgupta, S.; Delignat, S.; Marden, M.C.; Kaveri, S.V.; Lacroix-Desmazes, S. Heme binds to factor VIII and inhibits its interaction with activated factor IX. J. Thromb. Haemost. 2012, 10, 1062–1071. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Chantrathammachart, P.; Wang, S.; Jonas, W.; Kirchhofer, D.; Gailani, D.; Gruber, A.; Kasthuri, R.; Key, N.S.; Mackman, N.; et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica 2015, 100, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.L. Hematin-derived anticoagulant retards fibrin formation and is inhibited by iron chelators. Ann. N. Y. Acad. Sci. 1986, 485, 418–420. [Google Scholar] [CrossRef]
- Arkebauer, M.R.; Kanaparthy, S.S.; Malayaman, S.N.; Vosseller, K.; Nielsen, V.G. Carbon monoxide and nitric oxide modulate α2-antiplasmin and plasmin activity. Blood Coagul. Fibrinolysis 2011, 22, 712–719. [Google Scholar] [CrossRef]
- Wagener, F.A.D.T.G.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E-selectin in vascular endothelial cells. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Setty, B.N.Y.; Betal, S.G.; Zhang, J.; Stuart, M.J. Heme induces endothelial tissue factor expression: Potential role in hemostatic activation in patients with hemolytic anemia. J. Thromb. Haemost. 2008, 6, 2202–2209. [Google Scholar] [CrossRef] [Green Version]
- Rehani, T.; Mathson, K.; Belcher, J.D.; Vercellotti, G.M.; Slungaard, A. Heme potently stimulates tissue factor expression by peripheral blood monocytes: A novel mechanism for thrombosis in intravascular hemolytic diseases. Blood 2013, 122, 2215. [Google Scholar] [CrossRef]
- Souza, G.R.; Fiusa, M.M.L.; Lanaro, C.; Colella, M.P.; Montalvao, S.A.L.; Saad, S.T.O.; Costa, F.F.; Traina, F.; Annichino-Bizzacchi, J.M.; De Paula, E.V. Coagulation activation by heme: Evidence from global hemostasis assays. Blood 2014, 124, 455. [Google Scholar] [CrossRef]
- Hounkpe, B.W.; Moraes, C.R.P.; do Santos, M.N.N.; Costas, F.F.; De Paula, E.V. Heme induces mRNA expression and activation of tissue factor by TLR4 dependent mechanisms. medRxiv 2020. [Google Scholar] [CrossRef]
- May, O.; Yatime, L.; Merle, N.S.; Delguste, F.; Howsam, M.; Daugan, M.V.; Paul-Constant, C.; Billamboz, M.; Ghinet, A.; Lancel, S.; et al. The receptor for advanced glycation end products is a sensor for cell-free heme. FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Pierach, C.A.; Rosborough, T.K. Points: Malaria, haematin, and coagulation. Br. Med. J. 1985, 290, 793. [Google Scholar] [CrossRef] [Green Version]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil extracellular traps. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]
- Shet, A.S.; Lizarralde-Iragorri, M.A.; Naik, R.P. The molecular basis for the prothrombotic state in sickle cell disease. Haematologica 2020, 105, 2368–2379. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hopp, M.-T.; Imhof, D. Linking Labile Heme with Thrombosis. J. Clin. Med. 2021, 10, 427. https://doi.org/10.3390/jcm10030427
Hopp M-T, Imhof D. Linking Labile Heme with Thrombosis. Journal of Clinical Medicine. 2021; 10(3):427. https://doi.org/10.3390/jcm10030427
Chicago/Turabian StyleHopp, Marie-Thérèse, and Diana Imhof. 2021. "Linking Labile Heme with Thrombosis" Journal of Clinical Medicine 10, no. 3: 427. https://doi.org/10.3390/jcm10030427
APA StyleHopp, M.-T., & Imhof, D. (2021). Linking Labile Heme with Thrombosis. Journal of Clinical Medicine, 10(3), 427. https://doi.org/10.3390/jcm10030427