
Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: A Review of Potential Implications

 , ,
, ,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Main Text
2.1. Search Strategy and Selection Criteria
2.2. Quality of Anticoagulation with VKA
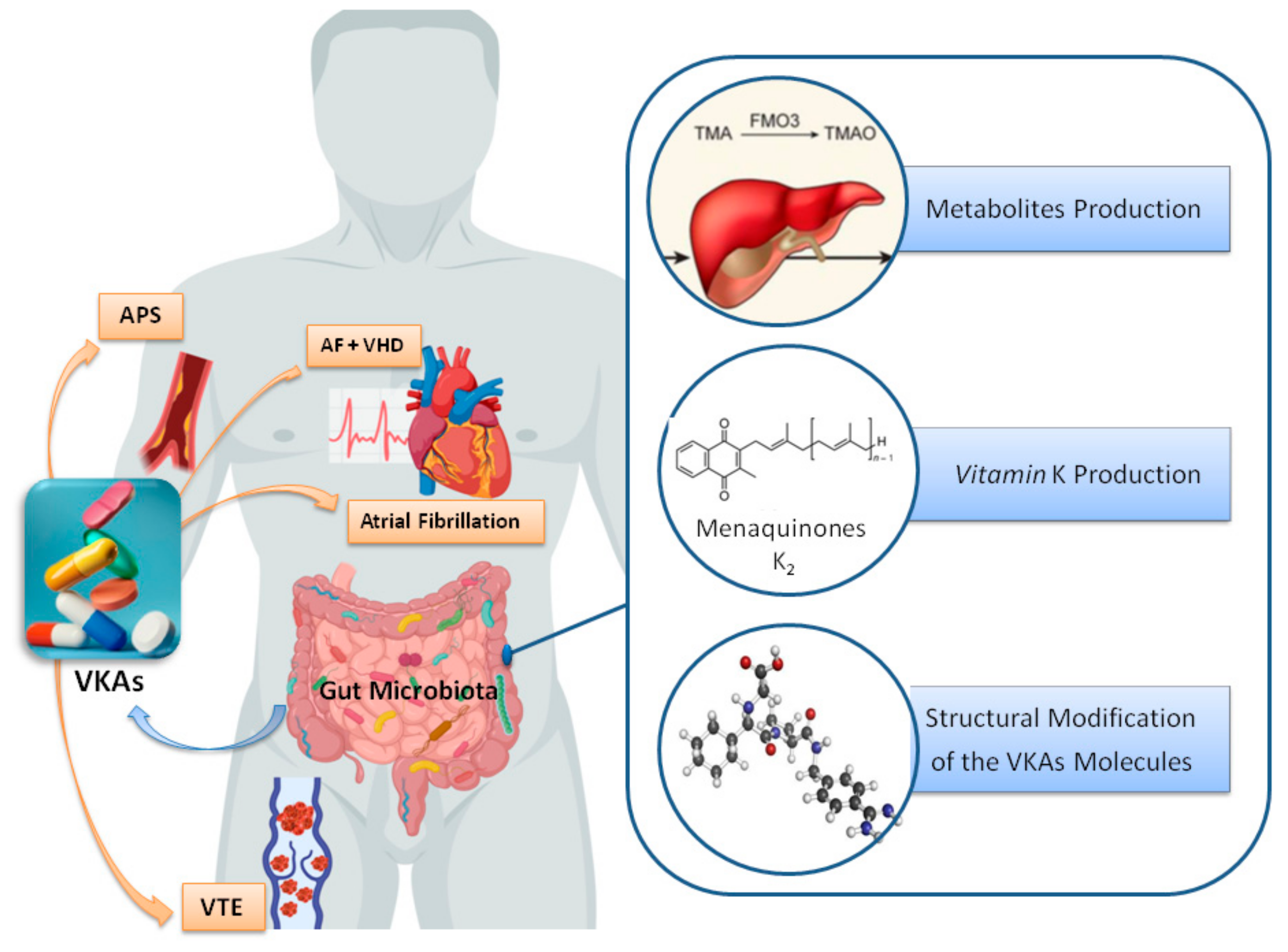
2.3. Gut Microbiota and Xenobiotic Metabolism
2.3.1. Effect of Metabolites Produced by Gut Microbiota on VKAs Drugs
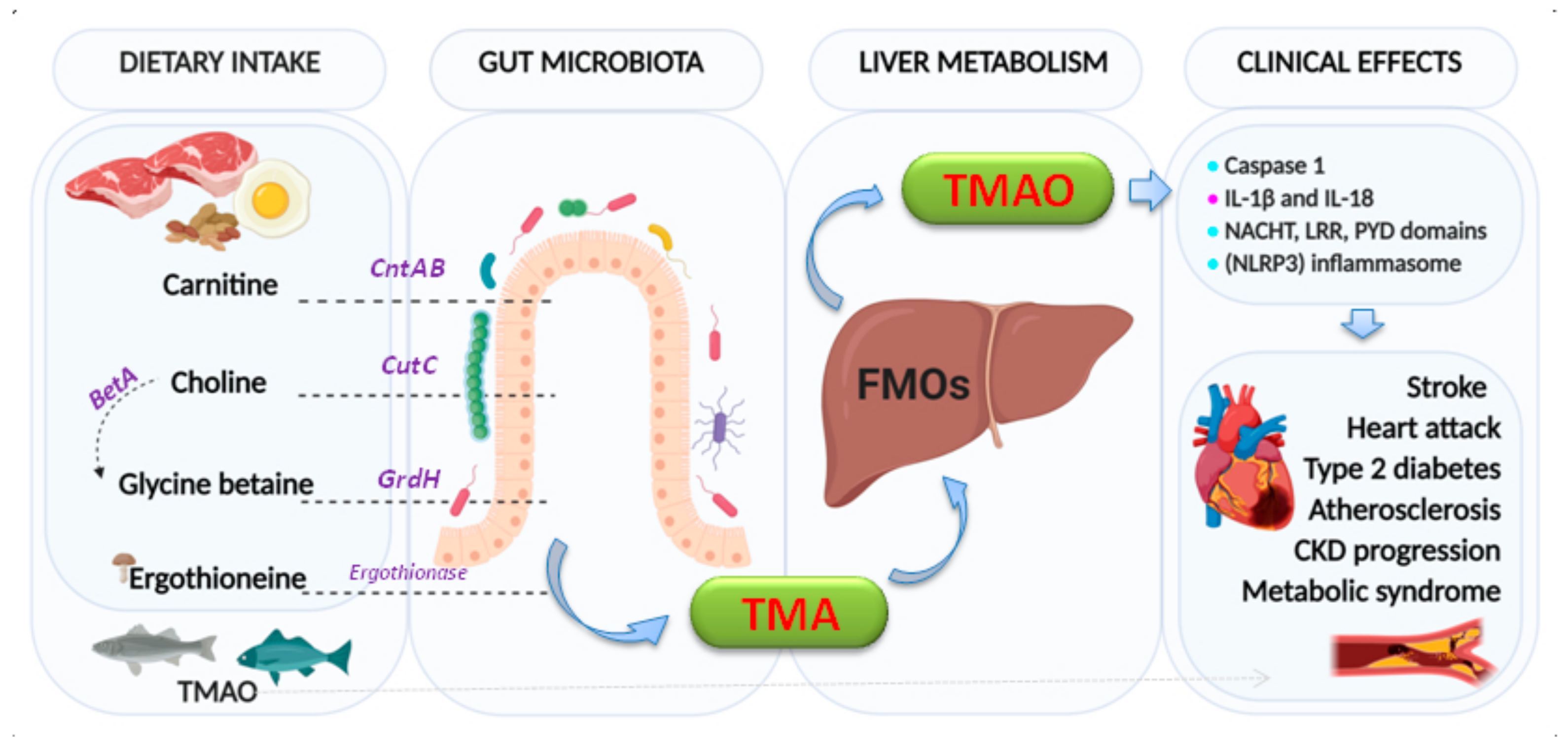
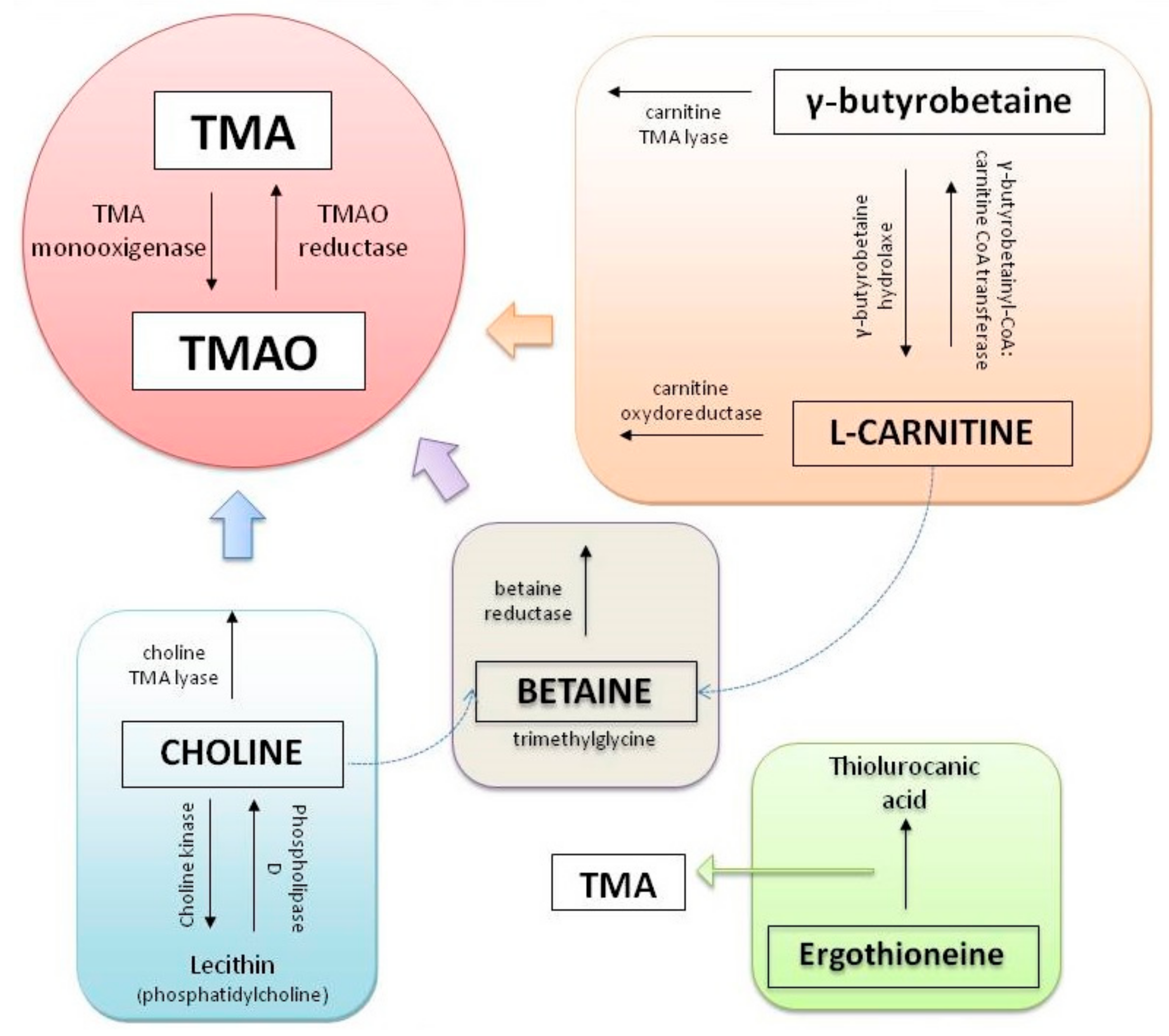
Trimethylamine N-Oxide (TMAO)
Indoxyl Sulfate (IS) and Indole-3 Acetic Acid (IAA)
2.3.2. Effect of Vitamin K-Producing Bacteria
2.3.3. Structural Modification of the VKAs Molecules by Gut Bacteria
2.4. Outstanding Questions
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APS | antiphospholipid syndrome |
| COX-2 | cyclooxygenase-2 |
| DOACs | direct-acting oral anticoagulants |
| Gas6 | growth arrest specific protein 6 |
| GBB | γ-butyrobetaine |
| GM | gut microbiota |
| GP | ganglionated plexi |
| GRP | gla-rich protein |
| HMGB1 | high mobility group box 1 |
| IAA | indole-3 acetic acid |
| INR | international normalized ratio |
| IS | indoxyl Sulfate |
| MGP | matrix Gla protein |
| NF-κB | nuclear factor-κB |
| OAC | oral anticoagulation |
| OACs | oral anticoagulants |
| Oc | osteocalcin |
| PLF | periostin-like-factor |
| PRGP | proline-richGla proteins |
| TMA | trimethylamine |
| TMAO | trimethylamine N-Oxide |
| TMG | transmembrane Gla protein |
| TNF-α | tumor necrosis factor-alpha |
| TTR | therapeutic range |
| VKAs | vitamin K antagonists |
| VKORC | vitamin K epoxide reductase |
| VKORC1 | vitamin K oxide reductasecomplex 1 |
| VTE | venous thromboembolism |
References
- McIntyre, W.F.; Healey, J.S. Stroke Prevention for Patients with Atrial Fibrillation: Beyond the Guidelines. J. Atr. Fibrillation. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Banerjee, A.; Boriani, G.; Chiang, C.E.; Fargo, R.; Freedman, B.; Lane, D.A.; Ruff, C.T.; Turakhia, M.; Werring, D.; et al. Antithrombotic Therapy for Atrial Fibrillation. Chest 2018, 154, 1121–1201. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Benussi, S.; Kotecha, D.; Ahlsson, A.; Atar, D.; Casadei, B.; Castella, M.; Diener, H.; Heidbuchel, H.; Hendriks, J.; et al. 2016 ESC Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur. Heart. J. 2016, 37, 2893–2962. [Google Scholar] [CrossRef]
- Boriani, G.; Proietti, M.; Laroche, C.; Fauchier, L.; Marin, F.; Nabauer, M.; Potpara, T.; Dan, G.-A.; Kalarus, Z.; Diemberger, I.; et al. Contemporary stroke prevention strategies in 11 096 European patients with atrial fibrillation: A report from the EURObservational Research Programme on Atrial Fibrillation (EORP-AF) Long-Term General Registry. Europace 2018, 20, 747–757. [Google Scholar] [CrossRef]
- Lip, G.Y.; Collet, J.P.; De Caterina, R.; Fauchier, L.; Lane, D.A.; Larsen, T.B.; Marin, F.; Morais, J.; Narasimhan, C.; Olshansky, B.; et al. Antithrombotic Therapy in Atrial Fibrillation Associated with Valvular Heart Disease: Executive Summary of a Joint Consensus Document from the European Heart Rhythm Association (EHRA) and European Society of Cardiology Working Group on Thrombosis, Endorsed by the ESC Working Group on Valvular Heart Disease, Cardiac Arrhythmia Society of Southern Africa (CASSA), Heart Rhythm Society (HRS), Asia Pacific Heart Rhythm Society (APHRS), South African Heart (SA Heart) Association and Sociedad Latinoamericana de EstimulaciónCardíaca y Electrofisiología (SOLEACE). Thromb. Haemost. 2017, 117, 2215–2236. [Google Scholar] [CrossRef]
- Marín, F.; Roldán, V. Optimizing Vitamin K Antagonist Treatment in Patients with Mechanical Heart Valve Prosthesis. Thromb. Haemost. 2018, 118, 806–807. [Google Scholar] [CrossRef]
- Pengo, V.; Denas, G.; Zoppellaro, G.; Jose, S.P.; Hoxha, A.; Ruffatti, A.; Andreoli, L.; Tincani, A.; Cenci, C.; Prisco, D.; et al. Rivaroxaban vs warfarin in high-risk patients with antiphospholipid syndrome. Blood 2018, 132, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Limper, M.; De Leeuw, K.; Lely, A.T.; Westerink, J.; Teng, Y.K.O.; Eikenboom, J.; Otter, S.; Jansen, A.J.G.; Ree, M.V.D.; Spierings, J.; et al. Diagnosing and treating antiphospholipid syndrome: A consensus paper. Neth. J. Med. 2019, 77, 98–108. [Google Scholar] [PubMed]
- Tritschler, T.; Castellucci, L.A. It’s time for head-to-head trials with direct oral anticoagulants. Thromb. Res. 2019, 180, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, T.; Czarnogorski, M.; Nizankowski, R.; Pilc, A. Are pharmacological properties of anticoagulants reflected in pharmaceutical pricing and reimbursement policy? Out-patient treatment of venous thromboembolism and utilization of anticoagulants in Poland. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1649–1656. [Google Scholar] [PubMed]
- Rivera-Caravaca, J.M.; Roldán, V.; Esteve-Pastor, M.A.; Valdés, M.; Vicente, V.; Marín, F.; Lip, G.Y. Reduced Time in Therapeutic Range and Higher Mortality in Atrial Fibrillation Patients Taking Acenocoumarol. Clin. Ther. 2018, 40, 114–122. [Google Scholar] [CrossRef]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.F.; Dwivedi, G.; O’Gara, F.; Caparros-Martin, J.; Ward, N.C. The gut microbiome and cardiovascular disease: Current knowledge and clinical potential. Am. J. Physiol. Circ. Physiol. 2019, 317, H923–H938. [Google Scholar] [CrossRef]
- Frankel, D.S.; Parker, S.E.; Rosenfeld, L.E.; Gorelick, P.B. HRS/NSA 2014 survey of atrial fibrillation and stroke: Gaps in knowledge and perspective, opportunities for improvement. Hear. Rhythm. 2015, 12, e105–e113. [Google Scholar] [CrossRef]
- Singer, D.E.; Hellkamp, A.S.; Piccini, J.P.; Mahaffey, K.W.; Lokhnygina, Y.; Pan, G.; Halperin, J.L.; Becker, R.C.; Breithardt, G.; Hankey, G.J.; et al. Impact of Global Geographic Region on Time in Therapeutic Range on Warfarin Anticoagulant Therapy: Data From the ROCKET AF Clinical Trial. J. Am. Hear. Assoc. 2013, 2, e000067. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Pastor, M.A.; Rivera-Caravaca, J.M.; Roldán-Rabadán, I.; Roldán, V.; Muñiz, J.; Raña-Míguez, P.; Ruiz-Ortiz, M.; Cequier, Á.; Bertomeu-Martinez, V.; Badimon, L.; et al. Quality of oral anticoagulation with vitamin K antagonists in ‘real-world’ patients with atrial fibrillation: A report from the prospective multicentre FANTASIIA registry. Europace 2018, 20, 1435–1441. [Google Scholar] [CrossRef]
- Holbrook, A.; Schulman, S.; Witt, D.M.; Vandvik, P.O.; Fish, J.; Kovacs, M.J.; Svensson, P.J.; Veenstra, D.L.; Crowther, M.; Guyatt, G.H. Evidence-based management of anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141 (Suppl. 2), e152S–e184S. [Google Scholar] [CrossRef] [PubMed]
- Barko, P.; McMichael, M.; Swanson, K.; Williams, D. The Gastrointestinal Microbiome: A Review. J. Veter. Intern. Med. 2017, 32, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nat. Cell Biol. 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; De Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the landscape of gut microbial community composition. Nat. Microbiol. 2018, 3, 8–16. [Google Scholar] [CrossRef]
- Zhong, H.; Penders, J.; Shi, Z.; Ren, H.; Cai, K.; Fang, C.; Ding, Q.; Thijs, C.; Blaak, E.E.; Stehouwer, C.D.A.; et al. Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in young school-age children. Microbiome 2019, 7, 1–14. [Google Scholar] [CrossRef]
- Krishnan, S.; Alden, N.; Lee, K. Pathways and functions of gut microbiota metabolism impacting host physiology. Curr. Opin. Biotechnol. 2015, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Doré, J.; Clement, K. The importance of the gut microbiota after bariatric surgery. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Patterson, A.D.; Idle, J.R.; Gonzalez, F.J. Xenobiotic Metabolomics: Major Impact on the Metabolome. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 37–56. [Google Scholar] [CrossRef]
- Swanson, H.I. Drug Metabolism by the Host and Gut Microbiota: A Partnership or Rivalry? Drug metabolism and disposition: The biological fate of chemicals. Drug Metab. Dispos. 2015, 43, 1499–1504. [Google Scholar] [CrossRef]
- Ioannides, C. Xenobiotic Metabolism: An Overview. In Enzyme Systems that Metabolise Drugs and Other Xenobiotics; Wiley: Hoboken, NJ, USA, 2002. [Google Scholar]
- Lake, B.G.; Price, R.J. Evaluation of the metabolism and hepatotoxicity of xenobiotics utilizing precision-cut slices. Xenobiotica 2013, 43, 41–53. [Google Scholar] [CrossRef]
- MacPherson, A.J.; Heikenwalder, M.; Ganal-Vonarburg, S.C. The Liver at the Nexus of Host-Microbial Interactions. Cell Host Microbe 2016, 20, 561–571. [Google Scholar] [CrossRef]
- Monga, S.P.S. (Ed.) Molecular Pathology of Liver Diseases; Springer: New York, NY, USA, 2011. [Google Scholar]
- Wang, B.H.L.; Siahaan, T. Drug Delivery: Principles and Applications; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Amedei, A.; Morbidelli, L. Circulating Metabolites Originating from Gut Microbiota Control Endothelial Cell Function. Molecules 2019, 24, 3992. [Google Scholar] [CrossRef] [PubMed]
- Mayneris-Perxachs, J.; Fernández-Real, J. Exploration of the microbiota and metabolites within body fluids could pinpoint novel disease mechanisms. FEBS J. 2019, 287, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Healey, G.; Murphy, R.; Brough, L.; Butts, C.A.; Coad, J. Interindividual variability in gut microbiota and host response to dietary interventions. Nutr. Rev. 2017, 75, 1059–1080. [Google Scholar] [CrossRef] [PubMed]
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The gastrointestinal microbiota as a site for the biotransformation of drugs. Int. J. Pharm. 2008, 363, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Galliher, P.M.; Cooney, C.L. Polysaccharide lyases. Appl. Biochem. Biotechnol. 1986, 12, 135–176. [Google Scholar] [CrossRef]
- Vernocchi, P.; Del Chierico, F.; Putignani, L. Gut Microbiota Metabolism and Interaction with Food Components. Int. J. Mol. Sci. 2020, 21, 3688. [Google Scholar] [CrossRef]
- Fung, T.C.; Olson, C.A.; Hsiao, T.C.F.C.A.O.E.Y. Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 2017, 20, 145–155. [Google Scholar] [CrossRef]
- Zimmermann, J.; Kaleta, C.; Waschina, S. gapseq: Informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Mar, M.-H.; Howe, J.C.; Holden, J.M. Concentrations of Choline-Containing Compounds and Betaine in Common Foods. J. Nutr. 2003, 133, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H. Choline deficiency. J. Nutr. Biochem. 1990, 1, 332–349. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Wishnok, J.S.; Blusztajn, J.K. Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 1983, 225, 320–324. [Google Scholar]
- Zeisel, S.H.; Warrier, M. TrimethylamineN-Oxide, the Microbiome, and Heart and Kidney Disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zheng, X.; Feng, M.; Li, D.; Zhang, H. Gut Microbiota-Dependent Metabolite Trimethylamine N-Oxide Contributes to Cardiac Dysfunction in Western Diet-Induced Obese Mice. Front. Physiol. 2017, 8, 139. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Zhu, X.H.; Ran, L.; Lang, H.D.; Yi, L.; Mi, M.T. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347. [Google Scholar] [CrossRef]
- Rohrmann, S.; Linseisen, J.; Allenspach, M.; Von Eckardstein, A.; Müller, D. Plasma Concentrations of Trimethylamine-N-oxide Are Directly Associated with Dairy Food Consumption and Low-Grade Inflammation in a German Adult Population. J. Nutr. 2016, 146, 283–289. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Yue, C.; Yang, X.; Li, J.; Chen, X.; Zhao, X.; Chen, Y.; Wen, Y. Trimethylamine N-oxide prime NLRP3 inflammasome via inhibiting ATG16L1-induced autophagy in colonic epithelial cells. Biochem. Biophys. Res. Commun. 2017, 490, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Heianza, Y.; Ma, W.; Manson, J.E.; Rexrode, K.M.; Qi, L. Gut Microbiota Metabolites and Risk of Major Adverse Cardiovascular Disease Events and Death: A Systematic Review and Meta-Analysis of Prospective Studies. J. Am. Hear. Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Lau, D.H.; Linz, D.; Schotten, U.; Mahajan, R.; Sanders, P.; Kalman, J.M. Pathophysiology of Paroxysmal and Persistent Atrial Fibrillation: Rotors, Foci and Fibrosis. Hear. Lung Circ. 2017, 26, 887–893. [Google Scholar] [CrossRef]
- Mishima, R.S.; Elliott, A.D.; Sanders, P.; Linz, D. Microbiome and atrial fibrillation. Int. J. Cardiol. 2018, 255, 103–104. [Google Scholar] [CrossRef]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.A.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nat. Cell Biol. 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Q.; Jiang, H. Gut microbiota in atherosclerosis: Focus on trimethylamine N-oxide. APMIS. 2020, 128, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Boukhlouf, S.; Fletcher, C. A bacterial metabolite, trimethylamine N-oxide, disrupts the hemostasis balance in human primary endothelial cells but no coagulopathy in mice. Blood Coagul. Fibrinolysis. 2019, 30, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Yoshida, M. Protein-Bound Uremic Toxins: New Culprits of Cardiovascular Events in Chronic Kidney Disease Patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef]
- Cipollone, F.; Cicolini, G.; Bucci, M. Cyclooxygenase and prostaglandin synthases in atherosclerosis: Recent insights and future perspectives. Pharmacol. Ther. 2008, 118, 161–180. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The Cardiovascular Effect of the Uremic Solute Indole-3 Acetic Acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2015, 27, 189–201. [Google Scholar] [CrossRef]
- Sorensen, A.B.; Tuneew, I.; Svensson, L.A.; Persson, E.; Østergaard, H.; Overgaard, M.T.; Olsen, O.H.; Gandhi, P.S. Beating tissue factor at its own game: Design and properties of a soluble tissue factor–independent coagulation factor VIIa. J. Biol. Chem. 2020, 295, 517–528. [Google Scholar] [CrossRef]
- Li, T.; Chang, C.-Y.; Jin, D.-Y.; Lin, P.-J.; Khvorova, A.; Stafford, D.W. Identification of the gene for vitamin K epoxide reductase. Nat. Cell Biol. 2004, 427, 541–544. [Google Scholar] [CrossRef]
- Ageno, W.; Gallus, A.S.; Wittkowsky, A.; Crowther, M.; Hylek, E.M.; Palareti, G. Oral anticoagulant therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012, 141 (Suppl. 2), e44S–e88S. [Google Scholar] [CrossRef]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [CrossRef]
- Wen, L.; Chen, J.; Duan, L.; Li, S. Vitamin K-dependent proteins involved in bone and cardiovascular health (Review). Mol. Med. Rep. 2018, 18, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Schurgers, L.J.; Uenishi, K. Comparison of menaquinone-4 and menaquinone-7 bioavailability in healthy women. Nutr. J. 2012, 11, 93. [Google Scholar] [CrossRef]
- Cosmi, B.; Palareti, G. Bleeding with anticoagulation therapy—Who is at risk, and how best to identify such patients. Thromb. Haemost. 2009, 102, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine, Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- Bolton-Smith, C.; Price, R.J.; Fenton, S.T.; Harrington, D.J.; Shearer, M.J. Compilation of a provisional UK database for the phylloquinone (vitamin K1) content of foods. Br. J. Nutr. 2000, 83, 389–399. [Google Scholar]
- Nimptsch, K.; Rohrmann, S.; Linseisen, J. Dietary intake of vitamin K and risk of prostate cancer in the Heidelberg cohort of the European Prospective Investigation into Cancer and Nutrition (EPIC-Heidelberg). Am. J. Clin. Nutr. 2008, 87, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Hojo, K.; Watanabe, R.; Mori, T.; Taketomo, N. Quantitative Measurement of Tetrahydromenaquinone-9 in Cheese Fermented by Propionibacteria. J. Dairy Sci. 2007, 90, 4078–4083. [Google Scholar] [CrossRef]
- Ichihashi, T.; Takagishi, Y.; Uchida, K.; Yamada, H. Colonic Absorption of Menaquinone-4 and Menaquinone-9 in Rats. J. Nutr. 1992, 122, 506–512. [Google Scholar] [CrossRef]
- Fujita, K.; Kakuya, F.; Ito, S. Vitamin K1 and K2 status and faecal flora in breast fed and formula fed 1-month-old infants. Eur. J. Nucl. Med. Mol. Imaging 1993, 152, 852–855. [Google Scholar] [CrossRef]
- Ramotar, K.; Conly, J.M.; Chubb, H.; Louie, T.J. Production of Menaquinones by Intestinal Anaerobes. J. Infect. Dis. 1984, 150, 213–218. [Google Scholar] [CrossRef]
- Beulens, J.W.J.; Booth, S.L.; Heuvel, E.G.H.M.V.D.; Stoecklin, E.; Baka, A.; Vermeer, C. The role of menaquinones (vitamin K2) in human health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Shearer, M.J.; Hamulyák, K.; Söcklin, E.; Vermeer, C. Effect of vitamin K intake on the stability of oral anticoagulant treatment: Dose-response relationships in healthy subjects. Blood 2004, 104, 2682–2689. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schurgers, L.J.; Teunissen, K.J.F.; Hamulyák, K.; Knapen, M.H.J.; Vik, H.; Vermeer, C. Vitamin K–containing dietary supplements: Comparison of synthetic vitamin K1 and natto-derived menaquinone-7. Blood 2006, 109, 3279–3283. [Google Scholar] [CrossRef] [PubMed]
- Cooke, G.; Behan, J.; Costello, M. Newly identified vitamin K-producing bacteria isolated from the neonatal faecal flora. Microb. Ecol. Health Dis. 2006, 18, 133–138. [Google Scholar] [CrossRef][Green Version]
- Obach, R.S. Pharmacologically Active Drug Metabolites: Impact on Drug Discovery and Pharmacotherapy. Pharmacol. Rev. 2013, 65, 578–640. [Google Scholar] [CrossRef]
- Guthrie, L.; Kelly, L. Bringing microbiome-drug interaction research into the clinic. EBioMedicine 2019, 44, 708–715. [Google Scholar] [CrossRef]
- Wilson, I.D.; Nicholson, J.K. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl. Res. 2017, 179, 204–222. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, J.; Jia, W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin. Drug Metab. Toxicol. 2016, 12, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.; MacGregor, A.; Carding, S.R. Drug-microbiota interactions and treatment response: Relevance to rheumatoid arthritis. AIMS Microbiol. 2018, 4, 642–654. [Google Scholar] [CrossRef]
- Rafii, F.; Cerniglia, C.E. Reduction of azo dyes and nitroaromatic compounds by bacterial enzymes from the human intestinal tract. Environ. Heal. Perspect. 1995, 103, 17–19. [Google Scholar] [CrossRef]
- Rafii, F.; Franklin, W.; Cerniglia, C.E. Azoreductase activity of anaerobic bacteria isolated from human intestinal microflora. Appl. Environ. Microbiol. 1990, 56, 2146–2151. [Google Scholar] [CrossRef] [PubMed]
- Lavrijsen, K.; Van Dyck, D.; Van Houdt, J.; Hendrickx, J.; Monbaliu, J.; Woestenborghs, R.; Meuldermans, W.; Heykants, J. Reduction of the prodrug loperamide oxide to its active drug loperamide in the gut of rats, dogs, and humans. Drug Metab. Dispos. 1995, 23, 354–362. [Google Scholar]
- Sharma, A.; Purkait, B. Identification of Medicinally Active Ingredient in Ultradiluted Digitalis purpurea: Fluorescence Spectroscopic and Cyclic-Voltammetric Study. J. Anal. Methods Chem. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Haiser, H.J.; Gootenberg, D.B.; Chatman, K.; Sirasani, G.; Balskus, E.P.; Turnbaugh, P.J. Predicting and Manipulating Cardiac Drug Inactivation by the Human Gut Bacterium Eggerthellalenta. Science 2013, 341, 295–298. [Google Scholar] [CrossRef]
- Matuskova, Z.; Anzenbacherova, E.; Vecera, R.; Tlaskalova-Hogenova, H.; Kolar, M.; Anzenbacher, P. Administration of a Probiotic Can Change Drug Pharmacokinetics: Effect of E. coli Nissle 1917 on Amidarone Absorption in Rats. PLoS ONE 2014, 9, e87150. [Google Scholar] [CrossRef]
- Zimmermann, M.; Zimmermann-Kogadeeva, M.; Wegmann, R.; Goodman, A.L. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nat. Cell Biol. 2019, 570, 462–467. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Liu, X.; Xiang, M.; Zhou, L.; Huang, C.; Shen, Z.; Miao, L.-Y. The gut microbes, Enterococcus and Escherichia-Shigella, affect the responses of heart valve replacement patients to the anticoagulant warfarin. Pharmacol. Res. 2020, 159, 104979. [Google Scholar] [CrossRef] [PubMed]
- Din, A.U.; Hassan, A.; Zhu, Y.; Yin, T.; Gregersen, H.; Wang, G. Amelioration of TMAO through probiotics and its potential role in atherosclerosis. Appl. Microbiol. Biotechnol. 2019, 103, 9217–9228. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Tao, X.; Xiong, H.; Yu, J.; Wei, H. Lactobacillus plantarum ZDY04 exhibits a strain-specific property of lowering TMAO via the modulation of gut microbiota in mice. Food Funct. 2018, 9, 4299–4309. [Google Scholar] [CrossRef] [PubMed]

 : activation,
: activation,  : producing and secretion.
: activation, : producing and secretion.
: producing and secretion.
: activation, : producing and secretion.


{kind=link}
{kind=link}
{kind=link}
| Co-Medication |
| Antiplatelet drugs |
| Drugs affecting pharmacokinetics or pharmacodynamics of VKAs |
| Non-steroidal anti-inflammatory drugs |
| Comorbidities |
| Cancer |
| Congestive heart failure |
| Liver diseases |
| History of atherosclerotic stroke |
| History of major bleeding |
| Uncontrolled hypertension |
| Genetic Factors |
| Mutation in factor IX propeptide (low factor IX levels) |
| Polymorphisms of VKORC1 and CYP2C9 |
| Natural Conditions |
| Advanced age |
| Female sex |
| Personal Characteristics/Life Habits |
| Absence of familiar or social support |
| Alcohol abuse |
| Insufficient information and education to the treatment |
| Nutritional supplements and herbal products |
| Poor compliance |
| Poor dietary intake of vitamin K |
| Tendency to falls |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camelo-Castillo, A.; Rivera-Caravaca, J.M.; Orenes-Piñero, E.; Ramírez-Macías, I.; Roldán, V.; Lip, G.Y.H.; Marín, F. Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: A Review of Potential Implications. J. Clin. Med. 2021, 10, 715. https://doi.org/10.3390/jcm10040715
Camelo-Castillo A, Rivera-Caravaca JM, Orenes-Piñero E, Ramírez-Macías I, Roldán V, Lip GYH, Marín F. Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: A Review of Potential Implications. Journal of Clinical Medicine. 2021; 10(4):715. https://doi.org/10.3390/jcm10040715
Chicago/Turabian StyleCamelo-Castillo, Anny, José Miguel Rivera-Caravaca, Esteban Orenes-Piñero, Inmaculada Ramírez-Macías, Vanessa Roldán, Gregory Y. H. Lip, and Francisco Marín. 2021. "Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: A Review of Potential Implications" Journal of Clinical Medicine 10, no. 4: 715. https://doi.org/10.3390/jcm10040715
APA StyleCamelo-Castillo, A., Rivera-Caravaca, J. M., Orenes-Piñero, E., Ramírez-Macías, I., Roldán, V., Lip, G. Y. H., & Marín, F. (2021). Gut Microbiota and the Quality of Oral Anticoagulation in Vitamin K Antagonists Users: A Review of Potential Implications. Journal of Clinical Medicine, 10(4), 715. https://doi.org/10.3390/jcm10040715