
Pearls and Pitfalls of Introducing Ketogenic Diet in Adult Status Epilepticus: A Practical Guide for the Intensivist

 , , and
, , and
Abstract
1. Introduction
2. Presentation
3. Question: How Is Prolonged Seizure Activity Classified and What Are Potential Etiologies to Be Considered?
4. Question: What Are the Initial Steps in the Therapeutic Algorithm for Status Epilepticus?
5. Question: What Are Potential Rescue Therapeutic Approaches to the Management of Super-Refractory Status Epilepticus?
6. Ketogenic Diet
7. Question: What Are Some Factors Should the Clinician Consider When Selecting and Initiating KD for Adults with SE?
Initiating KD Safely
8. Question: Should You Fast the Patient to Achieve Ketosis Quickly? If So, How Long and What Are Potential Consequences? If You Decide Not to Fast, Can Ketosis Still Be Achieved?
Variations in KD Protocols
9. Question: What Factors Can Impede the Success of Achieving Ketosis, and thus Jeopardize the Utility of KD?
10. Hidden Carbohydrates Can Hinder Achievement of Ketosis
11. Noncarbohydrate Related Hindrance of Ketosis
12. Question: Aside from Sedative Agents, What Other Widely Used Agents in the Neurological ICU Can Hinder Ketosis?
12.1. Antimicrobials and Respective Diluents as Source of Carbohydrates
12.2. Non “Medications” Contain Hidden Carbohydrates
12.3. Comprehensive Approach to Implementing KD in Adult SE
13. Question: What Laboratory Values Should the Intensivist Pay Particular Attention to When Using KD?
14. Ketosis Maintenance and Surveillance
15. Question: What Natural Physiologic Mechanisms Must Be Accounted for When Attempting to Achieve/Maintain Ketosis?
16. KD and Supplements
Question: What Supplements may Be Warranted when Starting a Ketogenic Diet?
17. Termination of Ketotic Therapy
Question: Once Anesthetics Have Been Weaned and/or Seizure Activity Has Improved, How Should KD Be Weaned?
18. Anticipating and Managing Complications
Question: What Are Some Potential Complications of KD?
19. Future of KD in Adult SE/RSE/SRSE/NORSE
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Cervenka, M.C.; Wood, S.; Bagary, M.; Balabanov, A.; Bercovici, E.; Brown, M.-G.; Devinsky, O.; Lorenzo, C.D.; Doherty, C.P.; Felton, E.; et al. International Recommendations for the Management of Adults Treated with Ketogenic Diet Therapies. Neurol. Clin. Pract. 2020. [Google Scholar] [CrossRef]
- Kaul, N.; Laing, J.; Nicolo, J.-P.; Nation, J.; Kwan, P.; O’Brien, T.J. Practical considerations for ketogenic diet in adults with super refractory status epilepticus. Neurol. Clin. Pract. 2020. [Google Scholar] [CrossRef]
- McDonald, T.J.W.; Cervenka, M.C. The Expanding Role of Ketogenic Diets in Adult Neurological Disorders. Brain Sci. 2018, 8, 148. [Google Scholar] [CrossRef]
- Thakur, K.T.; Probasco, J.C.; Hocker, S.E.; Roehl, K.; Henry, B.; Kossoff, E.H.; Kaplan, P.W.; Geocadin, R.G.; Hartman, A.L.; Venkatesan, A.; et al. Ketogenic diet for adults in super-refractory status epilepticus. Neurology 2014, 82, 665–670. [Google Scholar] [CrossRef]
- Cervenka, M.C.; Hocker, S.; Koenig, M.; Bar, B.; Henry-Barron, B.; Kossoff, E.H.; Hartman, A.L.; Probasco, J.C.; Benavides, D.R.; Venkatesan, A.; et al. Phase I/II multicenter ketogenic diet study for adult superrefractory status epilepticus. Neurology 2017, 88, 938–943. [Google Scholar] [CrossRef]
- Caraballo, R.H.; Flesler, S.; Armeno, M.; Fortini, S.; Agustinho, A.; Mestre, G.; Cresta, A.; Buompadre, M.C.; Escobal, N. Ketogenic diet in pediatric patients with refractory focal status epilepticus. Epilepsy Res. 2014, 108, 1912–1916. [Google Scholar] [CrossRef] [PubMed]
- Cobo, N.H.; Sankar, R.; Murata, K.K.; Sewak, S.L.; Kezele, M.A.; Matsumoto, J.H. The ketogenic diet as broad-spectrum treatment for super-refractory pediatric status epilepticus: Challenges in implementation in the pediatric and neonatal intensive care units. J. Child Neurol. 2015, 30, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Trinka, E.; Cock, H.; Hesdorffer, D.; Rossetti, A.O.; Scheffer, I.E.; Shinnar, S.; Shorvon, S.; Lowenstein, D.H. A definition and classification of status epilepticus—Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 2015, 56, 1515–1523. [Google Scholar] [CrossRef]
- Delgado-Escueta, A.V.; Wasterlain, C.; Treiman, D.M.; Porter, R.J. Status epilepticus: Summary. Adv. Neurol. 1983, 34, 537–541. [Google Scholar] [PubMed]
- Wasterlain, C.G.; Baxter, C.F.; Baldwin, R.A. GABA metabolism in the substantia nigra, cortex, and hippocampus during status epilepticus. Neurochem. Res. 1993, 18, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Yeh, J.L.; Kapur, J. Status epilepticus increases the intracellular accumulation of GABAA receptors. J. Neurosci. 2005, 25, 5511–5520. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Sun, C.; Yeh, J.L.; Mangan, P.S.; Kapur, J. GABA(A) receptor internalization during seizures. Epilepsia 2007, 48 (Suppl. 5), 109–113. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, H.P.; Joshi, S.; Mtchedlishvili, Z.; Brar, J.; Kapur, J. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 2008, 28, 2527–2538. [Google Scholar] [CrossRef] [PubMed]
- Brophy, G.M.; Bell, R.; Claassen, J.; Alldredge, B.; Bleck, T.P.; Glauser, T.; Laroche, S.M.; Riviello, J.J.; Riviello, J.J.; Shutter, L.; et al. Guidelines for the Evaluation and Management of Status Epilepticus. Neurocrit. Care 2012, 17, 3–23. [Google Scholar] [CrossRef]
- Hirsch, L.J.; Gaspard, N.; Baalen, A.v.; Nabbout, R.; Demeret, S.; Loddenkemper, T.; Navarro, V.; Specchio, N.; Lagae, L.; Rossetti, A.O.; et al. Proposed consensus definitions for new-onset refractory status epilepticus (NORSE), febrile infection-related epilepsy syndrome (FIRES), and related conditions. Epilepsia 2018, 59, 739–744. [Google Scholar] [CrossRef]
- Gaspard, N.; Foreman, B.P.; Alvarez, V.; Kang, C.C.; Probasco, J.C.; Jongeling, A.C.; Meyers, E.; Espinera, A.; Haas, K.F.; Schmitt, S.E.; et al. New-onset refractory status epilepticus: Etiology, clinical features, and outcome. Neurology 2015, 85, 1604–1613. [Google Scholar] [CrossRef]
- Leitinger, M.; Beniczky, S.; Rohracher, A.; Gardella, E.; Kalss, G.; Qerama, E.; Hofler, J.; Lindberg-Larsen, A.H.; Kuchukhidze, G.; Dobesberger, J.; et al. Salzburg Consensus Criteria for Non-Convulsive Status Epilepticus—Approach to clinical application. Epilepsy Behav. 2015, 49, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Sculier, C.; Gaspard, N. New onset refractory status epilepticus (NORSE). Seizure 2019, 68, 72–78. [Google Scholar] [CrossRef]
- Glauser, T.; Shinnar, S.; Gloss, D.; Alldredge, B.; Arya, R.; Bainbridge, J.; Bare, M.; Bleck, T.; Dodson, W.E.; Garrity, L.; et al. Evidence-Based Guideline: Treatment of Convulsive Status Epilepticus in Children and Adults: Report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016, 16, 48–61. [Google Scholar] [CrossRef]
- Chamberlain, J.M.; Kapur, J.; Shinnar, S.; Elm, J.; Holsti, M.; Babcock, L.; Rogers, A.; Barsan, W.; Cloyd, J.; Lowenstein, D.; et al. Efficacy of levetiracetam, fosphenytoin, and valproate for established status epilepticus by age group (ESETT): A double-blind, responsive-adaptive, randomised controlled trial. Lancet 2020, 395, 1217–1224. [Google Scholar] [CrossRef]
- Hawkes, M.A.; English, S.W.; Mandrekar, J.N.; Rabinstein, A.A.; Hocker, S. Causes of Death in Status Epilepticus. Crit. Care Med. 2019, 47, 1226–1231. [Google Scholar] [CrossRef] [PubMed]
- Caronna, E.; Vilaseca, A.; Gracia Gozalo, R.M.; Corral, A.S.; Santafe, M.; Sueiras, M.; Guzman, L.; Quintana, M.; Toledo-Argany, M.; Santamarina, E. Long-term prognosis related to deep sedation in refractory status Epilepticus. Acta Neurol. Scand. 2020, 142, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Prisco, L.; Ganau, M.; Aurangzeb, S.; Moswela, O.; Hallett, C.; Raby, S.; Fitzgibbon, K.; Kearns, C.; Sen, A. A pragmatic approach to intravenous anaesthetics and electroencephalographic endpoints for the treatment of refractory and super-refractory status epilepticus in critical care. Seizure 2020, 75, 153–164. [Google Scholar] [CrossRef]
- Ferlisi, M.; Hocker, S.; Trinka, E.; Shorvon, S. The anesthetic drug treatment of refractory and super-refractory status epilepticus around the world: Results from a global audit. Epilepsy Behav. 2019, 101, 106449. [Google Scholar] [CrossRef] [PubMed]
- Alkhachroum, A.; Der-Nigoghossian, C.A.; Mathews, E.; Massad, N.; Letchinger, R.; Doyle, K.; Chiu, W.-T.; Kromm, J.; Rubinos, C.; Velazquez, A.; et al. Ketamine to treat super-refractory status epilepticus. Neurology 2020, 95, e2286–e2294. [Google Scholar] [CrossRef]
- Trinka, E.; Leitinger, M. Which EEG patterns in coma are nonconvulsive status epilepticus? Epilepsy Behav. 2015, 49, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Vooturi, S.; Jayalakshmi, S.; Sahu, S.; Mohandas, S. Prognosis and predictors of outcome of refractory generalized convulsive status epilepticus in adults treated in neurointensive care unit. Clin. Neurol Neurosurg. 2014, 126, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Shorvon, S.; Ferlisi, M. The treatment of super-refractory status epilepticus: A critical review of available therapies and a clinical treatment protocol. Brain 2011, 134, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Tuzun, E.; Wu, H.-Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Annals Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef]
- Delft, R.v.; Lambrechts, D.; Verschuure, P.; Hulsman, J.; Majoie, M. Blood beta-hydroxybutyrate correlates better with seizure reduction due to ketogenic diet than do ketones in the urine. Seizure 2010, 19, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Cervenka, M.C. The role for ketogenic diets in epilepsy and status epilepticus in adults. Clin. Neurophysiol. Pract. 2017, 2, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Nabbout, R.; Mazzuca, M.; Hubert, P.; Peudennier, S.; Allarie, C.; Flurin, V.; Aberastury, M.; Silva, W.; Dulac, O. Efficacy of ketogenic diet in severe refractory status epilepticus initiating fever induced refractory epileptic encephalopathy in school age children (FIRES). Epilepsia 2010, 51, 2033–2037. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.H.; Ho-Huang, E.; Buhler, J. Systematic review of ketogenic diet use in adult patients with status epilepticus. Epilepsia Open 2020, 5, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Meira, I.D.; Romao, T.T.; Pires do Prado, H.J.; Kruger, L.T.; Paiva Pires, M.E.; da Conceicao, P.O. Ketogenic Diet and Epilepsy: What We Know So Far. Front. Neurosci. 2019, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Amark, P.E.; Ballaban-Gil, K.R.; Bergqvist, A.G.C.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009, 50, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Bergqvist, A.G.C.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- Baumeister, F.A.M.; Oberhoffer, R.; Liebhaber, G.M.; Kunkel, J.; Eberhardt, J.; Holthausen, H.; Peters, J. Fatal propofol infusion syndrome in association with ketogenic diet. Neuropediatrics 2004, 35, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, A.G.C.; Schall, J.I.; Gallagher, P.R.; Cnaan, A.; Stallings, V.A. Fasting versus Gradual Initiation of the Ketogenic Diet: A Prospective, Randomized Clinical Trial of Efficacy. Epilepsia 2005, 46, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Kang, H.C.; Park, J.C.; Kim, H.D. Benefits of the Nonfasting Ketogenic Diet Compared With the Initial Fasting Ketogenic Diet. Pediatrics 2004, 114, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Ford Flanel, D.E.A. The Yale-New Haven Hospital (Ynhh) Adult Nutritional Classification and Assessment Manual, 24th ed.; Yale New Haven Health System: New Haven, CT, August, 2017; pp. IV1–IV30. [Google Scholar]
- Zupec-Kania, B.; Abrahams, J.; Pietsch-Escuada, S. Charlie Foundation: White Paper Proceedings of Ketogenic Diet Therapies Symposium; The Charlie Foundation: Manhattan Beach, CA, USA, 2015. [Google Scholar]
- Arroliga, A.C.; Shehab, N.; McCarthy, K.; Gonzales, J.P. Relationship of continuous infusion lorazepam to serum propylene glycol concentration in critically ill adults*. Crit. Care Med. 2004, 32, 1709–1714. [Google Scholar] [CrossRef]
- Cameron, T. Carbohydrate Content of Medications. 28 July 2020 [cited 2020 December]. Available online: https://www.matthewsfriends.org/keto-therapies/keto-management/ketogenic-therapy-anti-epileptic-medications/carbohydrate-content-medications/ (accessed on 1 July 2020).
- Wilson, K.C.; Reardon, C.; Theodore, A.C.; Farber, H.W. Propylene Glycol Toxicity: A Severe Iatrogenic Illness in ICU Patients Receiving IV Benzodiazepines: A Case Series and Prospective, Observational Pilot Study. CHEST 2005, 128, 1674–1681. [Google Scholar] [CrossRef]
- Singh, S.; Khandelwal, A.; Datta, R.; Kaushal, A.; Singh, G.P. Recurrent Metabolic Acidosis during High-dose Midazolam Therapy for Refractory Status Epilepticus. Indian J. Crit. Care Med. Peer-Rev. Off. Publ. Indian Soc. Crit. Care Med. 2018, 22, 119–121. [Google Scholar]
- Reznik, M.E.; Berger, K.; Claassen, J. Comparison of Intravenous Anesthetic Agents for the Treatment of Refractory Status Epilepticus. J. Clin. Med. 2016, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Van Der Louw, E.J.T.M.; Desadien, R.; Vehmeijer, F.O.L.; van der Sijs, H.; Catsman-Berrevoets, C.E.; Neuteboom, R.F. Concomitant lamotrigine use is associated with decreased efficacy of the ketogenic diet in childhood refractory epilepsy. Seizure 2015, 32, 75–77. [Google Scholar] [CrossRef]
- Heo, G.; Kim, S.H.; Chang, M.J. Effect of ketogenic diet and other dietary therapies on anti-epileptic drug concentrations in patients with epilepsy. J. Clin. Pharm. Ther. 2017, 42, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Abulhasan, Y.B.; Rachel, S.P.; Chatillon-Angle, M.-O.; Alabdulraheem, N.; Schiller, I.; Dendukuri, N.; Angle, M.R.; Frenette, C. Healthcare-associated infections in the neurological intensive care unit: Results of a 6-year surveillance study at a major tertiary care center. Am. J. Infect. Control 2018, 46, 656–662. [Google Scholar] [CrossRef]
- Mcghee, B.; Katyal, N. Avoid Unnecessary Drug-Related Carbohydrates for Patients Consuming the Ketogenic Diet. J. Am. Diet. Assoc. 2001, 101, 87–101. [Google Scholar] [CrossRef]
- Munro, C.L.; Grap, M.J.; Jones, D.J.; McClish, D.K.; Sessler, C.N. Chlorhexidine, Toothbrushing, and Preventing Ventilator-Associated Pneumonia in Critically Ill Adults. Am. J. Crit. Care 2009, 18, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Zupec-Kania, B.; Foundation, C. Appendix 3: Minimising the Carbohydrate Content of Medications on Theketogenicdiet. Available online: https://media.gosh.nhs.uk/documents/APPENDIX_3_Minimising_the_carbohydrate_content_of_medications_on_the_ketogenic_diet.pdf (accessed on 1 August 2020).
- Zupec-Kania, B.; Zupanc, M.L. Long-term management of the ketogenic diet: Seizure monitoring, nutrition, and supplementation. Epilepsia 2008, 49, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Francis, B.A.; Fillenworth, J.; Gorelick, P.; Karanec, K.; Tanner, A. The Feasibility, Safety and Effectiveness of a Ketogenic Diet for Refractory Status Epilepticus in Adults in the Intensive Care Unit. Neurocrit. Care 2019, 30, 652–657. [Google Scholar] [CrossRef] [PubMed]
- David, B.; Mervyn, S. Hyperglycemia in Critical Illness: A Review. J. Diabetes Sci. Technol. 2009, 3, 1250–1260. [Google Scholar]
- Zupec-Kania, B.A.; Aldaz, V.; Montgomery, M.E.; Kostas, K.C. Enteral and Parenteral Applications of Ketogenic Diet Therapy. ICAN Infant Child Adolesc. Nutr. 2011, 3, 274–281. [Google Scholar] [CrossRef]
- Worden, L.T.; Turner, Z.; Pyzik, P.L.; Rubenstein, J.E.; Kossoff, E.H. Is there an ideal way to discontinue the ketogenic diet? Epilepsy Res. 2011, 95, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.C.; Pyzik, P.L.; Kossoff, E.H. Discontinuing the Ketogenic Diet in Seizure-Free Children: Recurrence and Risk Factors. Epilepsia 2007, 48, 187–190. [Google Scholar] [CrossRef]
- Kang, H.C.; Chung, D.E.; Kim, D.W.; Kim, H.D. Early- and Late-onset Complications of the Ketogenic Diet for Intractable Epilepsy. Epilepsia 2004, 45, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, K.; Watanabe, M.; Yamada, Y.; Shiihara, T. Protein-Losing Enteropathy as a Rare Complication of the Ketogenic Diet. Pediatric Neurol. 2015, 52, 526–528. [Google Scholar] [CrossRef] [PubMed]


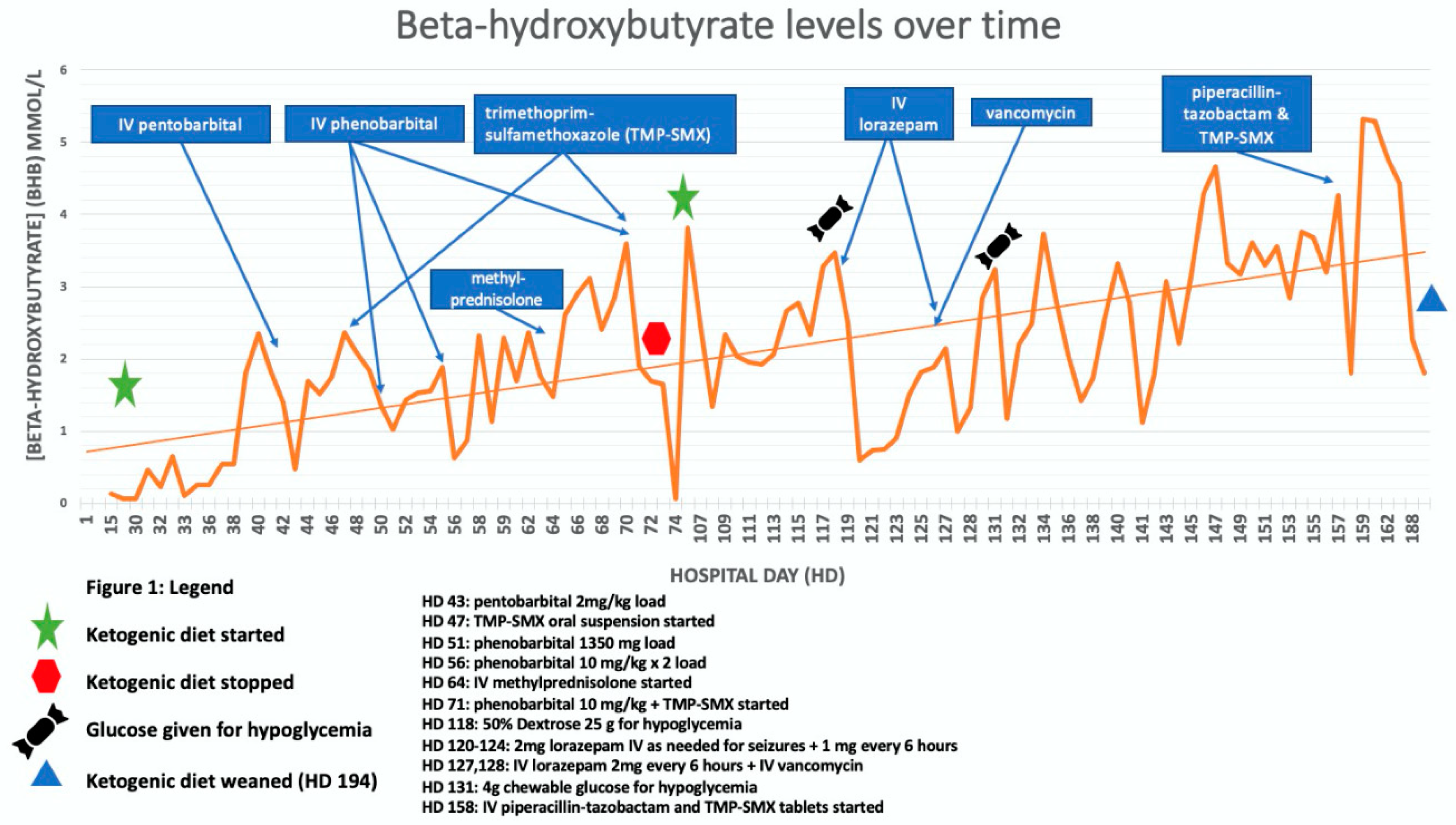{kind=link}
| Categories | Serum | CSF | Imaging | Pathology |
|---|---|---|---|---|
| Infectious: | Influenza A/B, H1N1, RPR, HIV, cat scratch panel, tick borne panel, Mycoplasma pneumonia, B Henselae, B quintana (all negative) | West Nile Virus, Enterovirus, Bacterial culture, HSV, VZV, Lyme disease, fungal culture, HHV6, EBV, Mycoplasma pneumoniae (all negative) | ||
| Inflammatory: | ANA, dsDNA, SSA, SSB, SCL 70, CRP, ESR, TPO antibody, thyroglobulin antibody, complements (C3, C4, CH50), ANCA, B2 glycoprotein, anticardiolipin, Antiribosomal P protein Ab, ACE, smooth muscle antibody, skeletal muscle antibody | AMPA-R Ab, CASPR2 Ab, DPPX Ab, GABA-B-R Ab, GAD 65, GFAP, LGI1-IgG, mGluR1 Ab, NMDA R Ab | ||
| Paraneoplastic: | GAD 65, NMDA, voltage gated potassium channel antibody, flow cytometry | AChR ganglionic neuronal Ab, Amphiphysin Ab, Antiglial nuclear Ab, Antineuronal nuclear Ab, CRMP-5, Neuronal (V-G) K+ channel Ab, N-Type Calcium channel ab, P/Q type calcium channel Ab, Purkinje cell cytoplasmic Ab, Striational Ab (all negative) | ||
| Metabolic: | TSH (0.22), Free T4 (1.7 ng/dL), Ammonia (47, 33, 37 µL/dL), serum and urine toxicology (negative) | |||
| MRI brain w/wo contrast: restricted diffusion and hyperintense FLAIR signal in the bilateral hippocampi | Benign ovarian cyst | |||
| CT Chest/abdomen/pelvis: no evidence of ovarian teratoma or other malignancy | No malignant cells in CSF | |||
| US pelvis: tiny 3–4 mm echogenic focus on the left ovary which may represent a small calcification, however, a tiny teratoma cannot be excluded | ||||
| MRI pelvis: no evidence of ovarian teratoma |
| Intravenous Product (General Product Concentration) | Carbohydrate Excipient and Amount Per Vial | Carbohydrate Content at a Common Dose | Fat Content | Alcohol Content |
| Brivaracetam (10 mg/mL) | – | – | – | – |
| Diazepam (5 mg/mL) [41] | Propylene glycol: 414 mg | 828 mg CHO/10 mg | – | 79 mg |
| Famotidine (10 mg/mL) [41] | Mannitol: 20 mg | 40 mg CHO/40 mg | – | – |
| Fosphenytoin [42] | – | – | – | – |
| Lorazepam (2 mg/mL) [42] | Propylene glycol: 753 mg | – | – | – |
| Pentobarbital (50 mg/mL) [41] | Propylene glycol: 414 mg | – | – | 79 mg |
| Phenobarbital (130 mg/mL) [41] | Propylene glycol: 702 mg | – | – | 79 mg |
| Phenytoin (50 mg/mL) [41] | Propylene glycol: 414 mg | – | – | 79 mg |
| Propofol (10 mg/mL) [41] | Glycerol: 22.5 mg/mL | 450 mg CHO/h (20 mL/h) | Soybean oil: 100 mg/mL | Benzyl alcohol * |
| Egg Lecithin: 12 mg/mL | ||||
| Lipid: 100 mg/mL (1.1 kcal/mL) | ||||
| Ketamine (multiple) | – | – | – | – |
| Lacosamide (multiple) | – | – | – | – |
| Midazolam (multiple) | – | – | – | Benzyl alcohol † |
| Thiopental (25 mg/mL) | – | – | – | – |
| Valproate (20 mg/mL) | – | – | – | – |
| Trimethoprim-sulfamethoxazole (Bactrim®) diluted in Dextrose 5% W 100 mL per 80–400 mg TMP-SMX | Dextrose: 5 g/100 ml | Up to 20 g CHO per dose | – | – |
| Vancomycin (Vancocin®) diluted in Dextrose 5% W per 1 g/250 mL solution | Dextrose: 5 g/100 ml | Up to 2 g CHO per dose | – | – |
| Enteral Product § (General Product Strength) | Carbohydrate Excipient and Amount Per Unit | Carbohydrate Content at a Common Dose | Fat Content | Alcohol Content |
| Carbamazepine (extended-release tablet) | Lactose monohydrate ‡ Microcrystalline cellulose ‡ | – | – | – |
| Clobazam (10 mg tablet) [43] | 105.3 mg/tablet | ≈100 mg/10 mg | – | – |
| Clonazepam (0.5 mg tablet) [43] | 143.5 mg/tablet | ≈2800 mg/10 mg | – | – |
| Levetiracetam (immediate release tablet) | Croscarmellose sodium ‡ Polyethylene glycol 3350 ‡ Polyethylene glycol 6000 ‡ | – | – | Polyvinyl alcohol |
| Psyllium (Metamucil) packet | 9 g CHO/tablespoon | 27 g CHO/day (TID) | – | – |
| Divalproex sodium (extended-release tablet) | Hypromelloses ‡ Lactose monohydrate ‡ Polyethylene glycol ‡ Propylene glycol ‡ Macrogol ‡ Microcrystalline cellulose ‡ | – | – | n-Butyl alcohol Isopropyl alcohol Polyvinyl alcohol |
| Pearls to Consider for Starting and Maintaining a Ketogenic Diet (KD) |
|---|
| I. KD initiation |
|
|
|
|
|
|
|
|
|
|
| II. KD maintenance |
|
| III. Pitfalls to consider: |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katz, J.B.; Owusu, K.; Nussbaum, I.; Beekman, R.; DeFilippo, N.A.; Gilmore, E.J.; Hirsch, L.J.; Cervenka, M.C.; Maciel, C.B. Pearls and Pitfalls of Introducing Ketogenic Diet in Adult Status Epilepticus: A Practical Guide for the Intensivist. J. Clin. Med. 2021, 10, 881. https://doi.org/10.3390/jcm10040881
Katz JB, Owusu K, Nussbaum I, Beekman R, DeFilippo NA, Gilmore EJ, Hirsch LJ, Cervenka MC, Maciel CB. Pearls and Pitfalls of Introducing Ketogenic Diet in Adult Status Epilepticus: A Practical Guide for the Intensivist. Journal of Clinical Medicine. 2021; 10(4):881. https://doi.org/10.3390/jcm10040881
Chicago/Turabian StyleKatz, Jason B., Kent Owusu, Ilisa Nussbaum, Rachel Beekman, Nicholas A. DeFilippo, Emily J. Gilmore, Lawrence J. Hirsch, Mackenzie C. Cervenka, and Carolina B. Maciel. 2021. "Pearls and Pitfalls of Introducing Ketogenic Diet in Adult Status Epilepticus: A Practical Guide for the Intensivist" Journal of Clinical Medicine 10, no. 4: 881. https://doi.org/10.3390/jcm10040881
APA StyleKatz, J. B., Owusu, K., Nussbaum, I., Beekman, R., DeFilippo, N. A., Gilmore, E. J., Hirsch, L. J., Cervenka, M. C., & Maciel, C. B. (2021). Pearls and Pitfalls of Introducing Ketogenic Diet in Adult Status Epilepticus: A Practical Guide for the Intensivist. Journal of Clinical Medicine, 10(4), 881. https://doi.org/10.3390/jcm10040881