
Genetics of Acromegaly and Gigantism


Abstract
:1. Introduction
2. Germline Mutations
2.1. GH Excess as an Isolated Pituitary Adenoma, FIPA
2.1.1. Aryl Hydrocarbon Receptor-Interacting Protein (AIP)
2.1.2. X-Linked Acrogigantism (XLAG)
2.2. Acromegaly as a Part of Systemic Disorder
2.2.1. Mutliple Endocrine Neoplasia Type 1 and Type 4 (MEN1 and MEN4)
2.2.2. McCune–Albright Syndrome (MAS)
2.2.3. Carney Complex (CNC)
2.2.4. Phaeochromocytoma/Paraganglioma (PPGL) and Pituitary Adenoma Association (3Pa)—SDHx/MAX Mutations
2.3. Other Syndromic Disease Associated with Germline Mutations and GH Excess without Visible Pituitary Tumour/Pituitary Hyperplasia
2.3.1. Neurofibromatosis Type 1 (NF1)
2.3.2. Deficiency of the Immunoglobulin Superfamily Member 1 (IGSF1)
2.3.3. Tuberous Sclerosis Complex (TSC)
3. Somatic Variants in GH-Secreting PitNETs
3.1. GNAS
3.2. Glucose-Dependent Insulinotropic Polypeptide Receptor (GIPR)
3.3. Other Genes
4. Recommendations for Genetic Screening in Acromegaly and Gigantism
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Melmed, S. Acromegaly. N. Engl. J. Med. 2006, 355, 2558–2573. [Google Scholar] [CrossRef]
- Kasuki, L.; da Silva Rocha, P.; Lamback, E.B.; Gadelha, M.R. Determinants of morbidities and mortality in acromegaly. Arch. Endocrinol. Metab. 2019, 63, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Maione, L.; Chanson, P. National acromegaly registries. Best. Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101264. [Google Scholar] [CrossRef]
- Gadelha, R.; Kasuki, L.; Lim, D.S.T.; Fleseriu, M. Systemic Complications of Acromegaly and the Impact of the Current Treatment Landscape: An Update. Endocr. Rev. 2018, 40, 268–332. [Google Scholar] [CrossRef] [Green Version]
- Lavrentaki, A.; Paluzzi, A.; Wass, J.A.H.; Karavitaki, N. Epidemiology of acromegaly: Review of population studies. Pituitary 2017, 20, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Asa, S.L.; Casar-Borota, O.; Chanson, P.; Delgrande, E.; Earls, P.; Ezzat, S.; Grossman, A.; Ikeda, H.; Inosshita, N.; Karavitaki, N.; et al. From pituitary adenoma to pituitary neuroendocrine tumor (pitnet): An international pituitary pathology club proposal. Endocr. Relat. Cancer 2017, 24, C5–C8. [Google Scholar] [CrossRef]
- Ho, K.K.Y.; Fleseriu, M.; Wass, J.; van der Lely, A.; Barkan, A.; Giustina, A.; Casanueva, F.F.; Heaney, A.P.; Biermasz, N.; Strasburger, C.; et al. The tale in evolution: Clarity, consistency and consultation, not contradiction and confusion. Pituitary 2020, 23, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Asa, S.L.; Asioli, S.; Bozkurt, S.; Casar-Borota, O.; Chinezu, L.; Comunoglu, N.; Cossu, G.; Cusimano, M.; Delgrange, E.; Earls, P.; et al. Pituitary neuroendocrine tumors (PitNETs): Nomenclature evolution, not clinical revolution. Pituitary 2020, 23, 322–325. [Google Scholar] [CrossRef]
- Ho, K.K.Y.; Fleseriu, M.; Wass, J.; van der Lely, A.; Barkan, A.; Giustina, A.; Casanueva, F.F.; Heaney, A.P.; Biermasz, N.; Srasburger, C.; et al. A tale of pituitary adenomas: To NET or not to NET: Pituitary Society position statement. Pituitary 2019, 22, 569–573. [Google Scholar] [CrossRef]
- Ho, K.; Fleseriu, M.; Kaiser, U.; Salvatori, R.; Brue, T.; Lopes, M.B.; Kunz, P.; Molitch, M.; Camper, S.A.; Gadelha, M.; et al. Pituitary Neoplasm Nomenclature Workshop: Does Adenoma Stand the Test of Time? J. Endocr. Soc. 2021. [Google Scholar] [CrossRef]
- Thorner, M.O.; Frohman, L.A.; Leong, D.A.; Thominet, J.; Downs, T.; Hellmann, P.; Chitwood, J.; Vaughan, J.M.; Vale, W. Extrahypothalamic Growth-Hormone-Releasing Factor (GRF) Secretion Is a Rare Cause of Acromegaly: Plasma GRF Levels in 177 Acromegalic Patients. J. Clin. Endocrinol. Metab. 1984, 59, 846–849. [Google Scholar] [CrossRef]
- Sala, E.; Ferrante, E.; Verrua, E.; Malchiodi, E.; Mantovani, G.; Filopanti, M.; Ferrero, S.; Pietrabissa, A.; Vanoli, A.; La Rosaa, S.; et al. Growth hormone-releasing hormone-producing pancreatic neuroendocrine tumor in a multiple endocrine neoplasia type 1 family with an uncommon phenotype. Eur. J. Gastroenterol. Hepatol. 2013, 25, 858–862. [Google Scholar] [CrossRef]
- Butler, P.W.; Cochran, C.S.; Merino, M.J.; Nguyen, D.M.; Schrump, D.S.; Gorden, P. Ectopic growth hormone-releasing hormone secretion by a bronchial carcinoid tumor: Clinical experience following tumor resection and long-Acting octreotide therapy. Pituitary 2012, 15, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Cambiaso, P.; Galassi, S.; Palmiero, M.; Mastronuzzi, A.; Del Bufalo, F.; Capolino, R.; Cacchione, A.; Buonuomo, A.S.; Gonfiantini, M.V.; Bartuli, A.; et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am. J. Med. Genet. Part A 2017, 173, 2353–2358. [Google Scholar] [CrossRef]
- Joustra, S.D.; Roelfsema, F.; Van Trotsenburg, A.S.P.; Schneider, H.J.; Kosilek, R.P.; Kroon, H.M.; Logan, J.G.; Butterfield, N.C.; Zhou, X.; Toufaily, C.; et al. IGSF1 Deficiency Results in Human and Murine Somatotrope Neurosecretory Hyperfunction. J. Clin. Endocrinol. Metab. 2020, 105, 70–84. [Google Scholar] [CrossRef]
- Rostomyan, L.; Daly, A.F.; Petrossians, P.; Nachev, E.; Lila, A.R.; Lecoq, A.L.; Lecumberri, B.; Trivellin, G.; Salvatori, R.; Moraitis, A.G.; et al. Clinical and genetic characterization of pituitary gigantism: An international collaborative study in 208 patients. Endocr. Relat. Cancer 2015, 22, 745–757. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Korbonits, M. Gigantism: X-linked acrogigantism and GPR101 mutations. Growth Horm. IGF Res. 2016, 30–31, 64–69. [Google Scholar] [CrossRef]
- Hernandez-Ramirez, L.C.; Gabrovska, P.; Denes, J.; Stals, K.; Trivellin, G.; Tilley, D.; Ferrau, F.; Evanson, J.; Ellard, S.; Grossman, A.B.; et al. Landscape of Familial Isolated and Young-Onset Pituitary Adenomas: Prospective Diagnosis in AIP Mutation Carriers. J. Clin. Endocrinol. Metab. 2015, 100, E1242–E1254. [Google Scholar] [CrossRef] [Green Version]
- Stratakis, C.A.; Kirschner, L.S.; Carney, J.A. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 2001, 86, 4041–4046. [Google Scholar] [CrossRef]
- Vergès, B.; Boureille, F.; Goudet, P.; Murat, A.; Beckers, A.; Sassolas, G.; Cougard, P.; Chambe, B.; Montvernay, C.; Calender, A. Pituitary Disease in MEN Type 1 (MEN1): Data from the France-Belgium MEN1 Multicenter Study. J. Clin. Endocrinol. Metab. 2002, 87, 457–465. [Google Scholar] [CrossRef]
- Pieterman, C.R.C.; de Laat, J.M.; Twisk, J.W.R.; van Leeuwaarde, R.S.; de Herder, W.W.; Dreijerink, K.M.A.; Hermus, A.R.M.M.; Dekkers, O.M.; van der Horst-Schrivers, A.N.A.; Drent, M.L.; et al. Long-term natural course of small nonfunctional pancreatic neuroendocrine tumors in MEN1-results from the Dutch MEN1 study group. J. Clin. Endocrinol. Metab. 2017, 102, 3795–3805. [Google Scholar] [CrossRef] [Green Version]
- Xekouki, P.; Szarek, E.; Bullova, P.; Giubellino, A.; Quezado, M.; Mastroyannis, S.A.; Mastorakos, P.; Wassif, C.A.; Raygada, M.; Rentia, N.; et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J. Clin. Endocrinol. Metab. 2015, 100, E710–E719. [Google Scholar] [CrossRef]
- Dénes, J.; Korbonits, M. The clinical aspects of pituitary tumour genetics. Endocrine 2021. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.; Korbonits, M. Update on the Genetics of Pituitary Tumors. Endocrinol. Metab. Clin. North Am. 2020, 49, 433–452. [Google Scholar] [CrossRef]
- Hannah-Shmouni, F.; Stratakis, C.A. An update on the genetics of benign pituitary adenomas in children and adolescents. Curr. Opin. Endocr. Metab. Res. 2018, 1, 19–24. [Google Scholar] [CrossRef]
- Gadelha, M.R.; Kasuki, L.; Korbonits, M. The genetic background of acromegaly. Pituitary 2017, 20, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.F.; Beckers, A. Familial Isolated Pituitary Adenomas (FIPA) and Mutations in the Aryl Hydrocarbon Receptor Interacting Protein (AIP) Gene. Endocrinol. Metab. Clin. North Am. 2015, 44, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.; Caimari, F.; Hernández-Ramírez, L.C.; Collier, D.; Iacovazo, D.; Ronaldson, A.; Magid, K.; Lim, C.T.; Stals, K.; Ellard, S.; et al. Significant benefits of AIP testing and clinical screening in familial isolated and young-onset pituitary tumors. J. Clin. Endocrinol. Metab. 2020, 105, e2247–e2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.F.; Rixhon, M.; Adam, C.; Dempegioti, A.; Tichomirowa, M.A.; Beckers, A. High prevalence of pituitary adenomas: A cross-sectional study in the province of Liège, Belgium. J. Clin. Endocrinol. Metab. 2006, 91, 4769–4775. [Google Scholar] [CrossRef]
- Carty, D.M.; Harte, R.; Drummond, R.S.; Ward, R.; Magid, K.; Collier, D.; Owens, M.; Korbonits, M. AIP variant causing familial prolactinoma. Pituitary 2020, 24, 48–52. [Google Scholar] [CrossRef]
- Daly, A.F.; Jaffrain-Rea, M.L.; Ciccarelli, A.; Valdes-Socin, H.; Rohmer, V.; Tamburrano, G.; Borson-Chazot, C.; Estour, B.; Ciccarelli, E.; Brue, T.; et al. Clinical characterization of familial isolated pituitary adenomas. J. Clin. Endocrinol. Metab. 2006, 91, 3316–3323. [Google Scholar] [CrossRef] [Green Version]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.H.; et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N. Engl. J. Med. 2014, 371, 2363. [Google Scholar] [CrossRef]
- Beckers, A.; Aaltonen, L.A.; Daly, A.F.; Karhu, A. Familial isolated pituitary adenomas (FIPA) and the Pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr. Rev. 2013, 34, 239–277. [Google Scholar] [CrossRef]
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliovaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J. Clin. Endocrinol. Metab. 2010, 95, E373–E383. [Google Scholar] [CrossRef] [Green Version]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tupputainen, K.; Ebeling, T.M.L.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef]
- Igreja, S.; Chahal, H.S.; King, P.; Bolger, G.B.; Srirangalingam, U.; Guasti, L.; Chapple, J.P.; Trivellin, G.; Gueorguiiev, M.; Guegan, K.; et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum. Mutat. 2010, 31, 950–960. [Google Scholar] [CrossRef] [Green Version]
- Leontiou, C.A.; Gueorguiev, M.; van der Spuy, J.; Quinton, R.; Lolli, F.; Hassan, S.; Chahal, H.S.; Igreja, S.C.; Jordan, S.; Rowe, J.; et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J. Clin. Endocrinol. Metab. 2008, 93, 2390–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, A.F.; Vanbellinghen, J.F.; Khoo, S.K.; Jaffrain-Rea, M.L.; Naves, L.A.; Guitelman, M.A.; Murat, A.; Emy, P.; Gimenez-Roqueplo, A.P.; Tamburrano, G.; et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: Analysis in 73 families. J. Clin. Endocrinol. Metab. 2007, 92, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.; Hunter, S.; Bradley, L.; Chahal, H.S.; Storr, H.L.; Akker, S.A.; Kumar, A.V.; Orme, S.M.; Evanson, J.; Abid, N.; et al. Clinical experience in the screening and management of a large kindred with familial isolated pituitary adenoma due to an aryl hydrocarbon receptor interacting protein (AIP) mutation. J. Clin. Endocrinol. Metab. 2014, 99, 1122–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, A.; Chahal, H.S.; Korbonits, M. Molecular genetics of the aip gene in familial pituitary tumorigenesis. Prog. Brain Res. 2010, 182, 229–253. [Google Scholar]
- Kasuki, L.; Neto, L.V.; Wildemberg, L.E.A.; Colli, L.M.; de Castro, M.; Takiya, C.M.; Gadelha, M.R. AIP expression in sporadic somatotropinomas is a predictor of the response to octreotide LAR therapy independent of SSTR2 expression. Endocr. Relat. Cancer 2012. [Google Scholar] [CrossRef] [Green Version]
- Kasuki, L.; Pinho, J.D.; Neto, V. Low Aryl Hydrocarbon Receptor-Interacting Protein Expression Is a Better Marker of Invasiveness in Somatotropinomas than Ki-67 and p53. Neuroendocrinology 2011, 913, 39–48. [Google Scholar] [CrossRef]
- Kuzhandaivelu, N.; Cong, Y.S.; Inouye, C.; Yang, W.M.; Seto, E. XAP2, a novel hepatitis B virus X-associated protein that inhibits X transactivation. Nucleic Acids Res. 1996, 24, 4741–4750. [Google Scholar] [CrossRef]
- Occhi, G.; Jaffrain-Rea, M.L.; Trivellin, G.; Albiger, N.; Ceccato, F.; de Menis, E.; Angelini, M.; Ferasin, S.; Beckers, A.; Mantero, F.; et al. The R304X mutation of the aryl hydrocarbon receptor interacting protein gene in familial isolated pituitary adenomas: Mutational hot-spot or founder effect? J. Endocrinol. Investig. 2010, 33, 800–805. [Google Scholar] [CrossRef] [Green Version]
- Salvatori, R.; Radian, S.; Diekmann, Y.; Iacovazzo, D.; David, A.; Gabrovska, P.; Grassi, G.; Bussell, A.M.; Stals, K.; Weber, A.; et al. In-frame seven amino-acid duplication in AIP arose over the last 3000 years, disrupts protein interaction and stability and is associated with gigantism. Eur. J. Endocrinol. 2017, 177, 257–266. [Google Scholar] [CrossRef]
- Chahal, H.S.; Stals, K.; Unterländer, M.; Balding, D.J.; Thomas, M.G.; Kumar, A.V.; Besser, G.M.; Atkinson, A.B.; Morrison, P.J.; Howlett, T.A.; et al. AIP mutation in pituitary adenomas in the 18th century and today. N. Engl. J. Med. 2011, 364, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Radian, S.; Diekmann, Y.; Gabrovska, P.; Holland, B.; Bradley, L.; Wallace, H.; Stals, K.; Bussell, A.M.; McGurren, K.; Cuesta, M.; et al. Increased Population Risk of AIP-Related Acromegaly and Gigantism in Ireland. Hum. Mutat. 2017, 38, 78–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivellin, G.; Korbonits, M. AIP and its interacting partners. J. Endocrinol. 2011, 210, 137–155. [Google Scholar] [CrossRef] [Green Version]
- Chahal, H.S.; Trivellin, G.; Leontiou, C.A.; Alband, N.; Fowkes, R.C.; Tahir, A.; Igreja, S.C.; Chapple, J.P.; Jordan, S.; Lupp, A.; et al. Somatostatin analogs modulate AIP in somatotroph adenomas: The role of the ZAC1 pathway. J. Clin. Endocrinol. Metab. 2012, 97, E1411–E1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadelha, M.R.; Kasuki, L.; Korbonits, M. Novel pathway for somatostatin analogs in patients with acromegaly. Trends Endocrinol. Metab. 2013, 24, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Tuominen, I.; Heliövaara, E.; Raitila, A.; Rautiainen, M.R.; Mehine, M.; Katainen, R.; Aittomaki, V.; Lehtonen, H.J.; Ahlsten, M.; Kivipelto, L.; et al. AIP inactivation leads to pituitary tumorigenesis through defective Gαi-cAMP signaling. Oncogene 2015, 34, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Hong, Y.; Xu, J.; Wu, Q.; Reis, C.; Yan, W.; Wang, W.; Zhang, J. A Novel Mutation of Aryl Hydrocarbon Receptor Interacting Protein Gene Associated with Familial Isolated Pituitary Adenoma Mediates Tumor Invasion and Growth Hormone Hypersecretion. World Neurosurg. 2019, 123, e45–e59. [Google Scholar] [CrossRef] [PubMed]
- Aflorei, E.D.; Klapholz, B.; Chen, C.; Radian, S.; Dragu, A.N.; Moderau, N.; Prodromou, C.; Ribeiro, P.S.; Stanewsky, R.; Korbonits, M. In vivo bioassay to test the pathogenicity of missense human AIP variants. J. Med. Genet. 2018, 55, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Bizzi, M.F.; Pinheiro, S.V.B.; Bolger, G.B.; Schweizer, J.R.d.O.L.; Giannetti, A.V.; Dang, M.N.; Ribeiro-Oliveira, A.; Korbonits, M. Reduced protein expression of the phosphodiesterases PDE4A4 and PDE4A8 in AIP mutation positive somatotroph adenomas. Mol. Cell Endocrinol. 2018, 476, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Reddy, K.S.; Rai, A.; Madugundu, A.K.; Solanki, H.S.; Bhansali, A.; Radotra, B.D.; Kumar, N.; Collier, D.; Iacovazzo, D.; et al. Surgery, Octreotide, Temozolomide, Bevacizumab, Radiotherapy, and Pegvisomant Treatment of an AIP Mutation‒Positive Child. J. Clin. Endocrinol. Metab. 2019, 104, 3539–3544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbonits, M.; Storr, H.; Kumar, A.V. Familial pituitary adenomas-Who should be tested for AIP mutations? Clin. Endocrinol. 2012, 77, 351–356. [Google Scholar] [CrossRef]
- Jaffrain-Rea, M.L.; Rotondi, S.; Turchi, A.; Occhi, G.; Barlier, A.; Peverelli, E.; Rostomyan, L.; Defilles, C.; Angelini, M.; Oliva, M.A.; et al. Somatostatin analogues increase AIP expression in somatotropinomas, irrespective of Gsp mutations. Endocr. Relat. Cancer 2013, 20, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Ezzat, S.; Caspar-Bell, G.M.; Chik, C.L.; Denis, M.C.; Dominque, M.E.; Imran, S.A.; Johnson, M.D.; Lochnan, H.A.; Nyomba, B.L.G.; Prebtani, A.; et al. Predictive markers for postsurgical medical management of acromegaly: A systematic review and consensus treatment guideline. Endocr. Pract. 2019, 25, 379–393. [Google Scholar] [CrossRef]
- Daly, A.F.; Rostomyan, L.; Betea, D.; Bonneville, J.F.; Villa, C.; Pellegata, N.S.; Waser, B.; Reubi, J.C.; Stephan, C.W.; Christ, E.; et al. Aip-mutated acromegaly resistant to first-generation somatostatin analogs: Long-term control with pasireotide lar in two patients. Endocr. Connect. 2019, 8, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Chiloiro, S.; Doglietto, F.; Trapasso, B.; Iacovazzo, D.; Giampietro, A.; di Nardo, F.; de Waure, C.; Lauriola, L.; Mangiola, A.; Anile, C.; et al. Typical and atypical pituitary adenomas: A single-center analysis of outcome and prognosis. Neuroendocrinology 2015, 101, 143–150. [Google Scholar] [CrossRef]
- Dénes, J.; Kasuki, L.; Trivellin, G.; Colli, L.M.; Takiya, C.M.; Stiles, C.E.; Barry, S.; de Castro, M.; Gadelha, M.R.; Korbonits, M. Regulation of aryl hydrocarbon receptor interacting protein (AIP) protein expression by MiR-34a in sporadic somatotropinomas. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Bogner, E.M.; Daly, A.F.; Gulde, S.; Karhu, A.; Irmler, M.; Beckers, J.; Mohr, H.; Beckers, A.; Pellegata, N.S. miR-34a is upregulated in AIP-mutated somatotropinomas and promotes octreotide resistance. Int. J. Cancer 2020, 147, 3523–3538. [Google Scholar] [CrossRef] [PubMed]
- Heck, A.; Ringstad, G.; Fougner, S.L.; Casar-Borota, O.; Nome, T.; Ramm-Pettersen, J.; Bollerslev, J. Intensity of pituitary adenoma on T2-weighted magnetic resonance imaging predicts the response to octreotide treatment in newly diagnosed acromegaly. Clin. Endocrinol. 2012, 77, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Fougner, S.L.; Casar-Borota, O.; Heck, A.; Berg, J.P.; Bollerslev, J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin. Endocrinol. 2012, 76, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckers, A.; Lodish, M.B.; Trivellin, G.; Rostomyan, L.; Lee, M.; Faucz, F.R.; Yuan, B.; Choong, C.S.; Caberg, J.H.; Verrua, E.; et al. X-linked acrogigantism syndrome: Clinical profile and therapeutic responses. Endocr. Relat. Cancer 2015, 22, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Naves, L.A.; Daly, A.F.; Dias, L.A.; Yuan, B.; Zakir, J.C.O.; Barra, G.B.; Palmeira, L.; Villa, C.; Trivellin, G.; Junior, A.J.; et al. Aggressive tumor growth and clinical evolution in a patient with X-linked acro-gigantism syndrome. Endocrine 2016, 51, 236–244. [Google Scholar] [CrossRef]
- Daly, A.F.; Lysy, P.A.; Desfilles, C.; Rostomyan, L.; Mohamed, A.; Caberg, J.H.; Raverot, V.; Castermans, E.; Marbaix, E.; Maiter, D.; et al. GHRH excess and blockade in X-LAG syndrome. Endocr. Relat. Cancer 2016, 23, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Iacovazzo, D.; Caswell, R.; Bunce, B.; Jose, S.; Yuan, B.; Hernández-Ramírez, L.C.; Kapur, S.; Caimari, F.; Evanson, J.; Ferrau, F.; et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol Commun. 2016, 4, 56. [Google Scholar] [CrossRef]
- Liang, H.; Gong, F.; Liu, Z.; Yang, Y.; Yao, Y.; Wang, R.; Wang, L.; Chen, M.; Pan, H.; Zhu, H. A Chinese case of X-linked acrogigantism and systemic review. Neuroendocrinology 2020. [Google Scholar] [CrossRef]
- Daly, A.F.; Yuan, B.; Fina, F.; Caberg, J.H.; Trivellin, G.; Rostomyan, L.; de Herder, W.W.; Naves, L.A.; Metzger, D.; Cuny, T.; et al. Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects. Endocr. Relat. Cancer 2016, 23, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.J.; Bell, J.; Chung, W.K.; David, R.; Oberfield, S.E.; Wardlaw, S.L. Childhood acromegaly due to X-linked acrogigantism: Long term follow-up. Pituitary 2016, 19, 560–564. [Google Scholar] [CrossRef]
- Rodd, C.; Millette, M.; Iacovazzo, D.; Stiles, C.E.; Barry, S.; Evanson, J.; Albrecht, S.; Caswell, R.; Bunce, B.; Josen, S.; et al. Somatic GPR101 duplication causing X-linked acrogigantism (XLAG)-Diagnosis and management. J. Clin. Endocrinol. Metab. 2016, 101, 1927–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise-Oringer, B.K.; Zanazzi, G.J.; Gordon, R.J.; Wardlaw, S.L.; Anyane-Yeboa, K.; Chung, W.K.; Kohn, B.; Wisoff, J.H.; David, R.; Oberfield, S.E. Familial X-Linked Acrogigantism: Postnatal Outcomes and Tumor Pathology in a Prenatally Diagnosed Infant and His Mother. J. Clin. Endocrinol. Metab. 2019, 104, 4667–4675. [Google Scholar] [CrossRef] [PubMed]
- Gläsker, S.; Vortmeyer, A.O.; Lafferty, A.R.A.; Hofman, P.L.; Li, J.; Weil, R.J.; Zhuang, Z.; Oldfield, E.H. Hereditary pituitary hyperplasia with infantile gigantism. J. Clin. Endocrinol. Metab. 2011, 96, E2078–E2087. [Google Scholar] [CrossRef] [Green Version]
- Moran, A.; Larson, R.; Kovacs, K.; Horvath, E.; Singer, W.; Sagman, U.; Reubi, J.C.; Wilson, C.B.; Pescovitz, O.H. Gigantism due to pituitary mammosomatotroph hyperplasia. N. Engl. J. Med. 1990, 323, 322–327. [Google Scholar] [CrossRef]
- Villa, C.; Lagonigro, M.S.; Magri, F.; Koziak, M.; Jaffrain-Rea, M.L.; Brauner, R.; Bouligand, J.; Junier, M.P.; di Rocco, F.; Sainte-Rose, C.; et al. Hyperplasia-adenoma sequence in pituitary tumorigenesis related to aryl hydrocarbon receptor interacting protein gene mutation. Endocr. Relat. Cancer 2011, 18, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L. MEN1 2012 Guideline. J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, L.; Guo, X.; Wang, Z.; Lian, W.; Deng, K.; Lu, L.; Xing, B.; Zhu, H. Pituitary adenomas in patients with multiple endocrine neoplasia type 1: A single-center experience in China. Pituitary 2019, 22, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Corbetta, S.; Pizzocaro, A.; Peracchi, M.; Beck-Peccoz, P.; Faglia, G.; Spada, A. Multiple endocrine neoplasia type 1 in patients with recognized pituitary tumours of different types. Clin. Endocrinol. 1997, 47, 507–512. [Google Scholar] [CrossRef]
- Cuny, T.; Pertuit, M.; Sahnoun-Fathallah, M.; Daly, A.; Occhi, G.; Odu, M.F.; Tabarin, A.; Nunes, M.L.; Delemer, B.; Rohmer, V.; et al. Genetic analysis in young patients with sporadic pituitary macroadenomas: Besides AIP don’t forget MEN1 genetic analysis. Eur. J. Endocrinol. 2013, 168, 533–541. [Google Scholar] [CrossRef] [Green Version]
- Nachtigall, L.B.; Guarda, F.J.; Lines, K.E.; Ghajar, A.; Dichtel, L.; Mumbach, G.; Zhao, W.; Zhang, X.; Tritos, N.A.; Swearingen, B.; et al. Clinical MEN-1 Among a Large Cohort of Patients With Acromegaly. J. Clin. Endocrinol. Metab. 2020, 105, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.C.; Roman, S.A.; Sosa, J.A. Parathyroidectomy in the elderly: Analysis of 7313 patients. J. Surg. Res. 2011, 170, 240–246. [Google Scholar] [CrossRef]
- Kamenický, P.; Mazziotti, G.; Lombès, M.; Giustina, A.; Chanson, P. Growth hormone, insulin-like growth factor-1, and the kidney: Pathophysiological and clinical implications. Endocr. Rev. 2014, 35, 234–281. [Google Scholar] [CrossRef] [Green Version]
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol. Cell Endocrinol. 2014, 386, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Marques, P.; Magalhães, D.; Caimari, F.; Hernández-Ramírez, L.C.; Collier, D.; Stals, K.; Ellard, S.; Drue, M.; Akker, S.; Waterhouse, M.; et al. Phenotypic differences between patients with familial pituitary neuroendocrine tumours due to MEN1 or AIP mutations. Endocr. Abstr. 2020. [Google Scholar] [CrossRef]
- Trouillas, J.; Labat-Moleur, F.; Sturm, N.; Kujas, M.; Heymann, M.F.; Figarella-Branger, D.; Patey, M.; Mazucca, M.; Decullier, E.; Verges, B.; et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am. J. Surg. Pathol. 2008, 32, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Gomez-Hernandez, K.; Kucharczyk, W.; Ridout, R.; Zadeh, G.; Gentili, F.; Ezzat, S.; Asa, S.L. Silent subtype 3 pituitary adenomas are not always silent and represent poorly differentiated monomorphous plurihormonal Pit-1 lineage adenomas. Mod. Pathol. 2016, 29, 131–142. [Google Scholar] [CrossRef]
- Borson-Chazot, F.; Garby, L.; Raverot, G.; Claustrat, F.; Raverot, V.; Sassolas, G. Acromegaly induced by ectopic secretion of GHRH: A review 30 years after GHRH discovery. Ann. Endocrinol. 2012, 73, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Isailovic, T.; Todorovic, V.; Milicevic, I.; Petakov, M.; Macut, D.; Ognjanovic, S.; Elezovic, V.; Skender-Gazibara, M.; popovic, B.; Antic, I.B.; et al. Ectopic co-secretion of growth hormone and growth hormone-releasing hormone from a neuroendocrine lung tumor in a patient with MEN1 syndrome. Endocr. Abstr. 2014. [Google Scholar] [CrossRef]
- Nadhamuni, V.S.; Iacovazzo, D.; Evanson, J.; Trouillas, J.; Kurzawinski, T.; Bhattacharya, S.; Korbonits, M. Unusual cause of gigantism-Growth hormone releasing hormone (GHRH)-secreting pancreatic neuroendocrine tumour in a patient with multiple endocrine neoplasia type 1 (MEN1). Endocr. Abstr. 2019. [Google Scholar] [CrossRef]
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1). Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Yarman, S.; Tuncer, F.N.; Serbest, E. Three Novel MEN1 Variants in AIP-Negative Familial Isolated Pituitary Adenoma Patients. Pathobiology 2019, 86, 128–134. [Google Scholar] [CrossRef]
- de Laat, J.M.; Dekkers, O.M.; Pieterman, C.R.C.; Kluijfhout, W.P.; Hermus, A.R.; Pereira, A.M.; van der Horst-Schrivers, A.N.; Drent, L.M.; Bisschop, P.H.; Havekes, B.; et al. Long-Term Natural Course of Pituitary Tumors in Patients With MEN1: Results From the Dutch MEN1 Study Group (DMSG). J. Clin. Endocrinol. Metab. 2015, 100, 3288–3296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmens, I.; van de ven Wim, J.M.; Kas, K.; Zhang, C.X.; Giraud, S.; Wautot, V.; Buisson, N.; de Witte, K.; Salandre, J.; Lenoir, G.; et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Hum. Mol. Genet. 1997, 6, 1177–1183. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type. Science 1997, 276, 404–406. [Google Scholar] [CrossRef]
- Concolino, P.; Costella, A.; Capoluongo, E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet. 2016, 209, 36–41. [Google Scholar] [CrossRef]
- Lemos, M.C.; Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2008, 29, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Beijers, H.J.B.H.; Stikkelbroeck, N.M.L.; Mensenkamp, A.R.; Pfundt, R.; van der Luijt, R.B.; Timmers, H.J.L.M.; Hermus, A.R.M.M.; Kempers, M.J.E. Germline and somatic mosaicism in a family with multiple endocrine neoplasia type 1 (MEN1) syndrome. Eur. J. Endocrinol. 2019, 180, K15–K19. [Google Scholar] [CrossRef] [PubMed]
- Mauchlen, R.; Carty, D.; Talla, M.; Drummond, R. Multiple endocrine neoplasia type 1 (MEN1) mosaicism caused by a c.124G>A variant in the MEN1 gene. Endocr. Abstr. 2019. [Google Scholar] [CrossRef]
- Thakker, R.V. Genetics of parathyroid tumours. J. Intern Med. 2016, 280, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef] [Green Version]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbis, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef] [Green Version]
- Cavaco, B.M.; Domingues, R.; Bacelar, M.C.; Cardoso, H.; Barros, L.; Gomes, L.; Ruas, M.M.A.; Agapito, A.; Garrao, A.; Pannett, A.A.J.; et al. Mutational analysis of Portuguese families with multiple endocrine neoplasia type 1 reveals large germline deletions. Clin. Endocrinol. 2002, 56, 465. [Google Scholar] [CrossRef] [PubMed]
- Mohr, H.; Pellegata, N.S. Animal models of MEN. Endocr. Relat. Cancer 2017, 24, T161–T177. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.K. Exploring the tumors of multiple endocrine neoplasia type 1 in mouse models for basic and preclinical studies. Int. J. Endocr. Oncol. 2014, 1, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsisaari, N.; Ross, J.; Wu, X.; Kowanetz, M.; Pal, N.; Hall, L.; Eastham-Anderson, J.; Forrest, W.F.F.; van Bruggen, N.; Peale, F.V.; et al. Blocking vascular endothelial growth factor-A inhibits the growth of pituitary adenomas and lowers serum prolactin level in a mouse model of multiple endocrine neoplasia type. Clin. Cancer Res. 2008, 14, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Höfler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef] [Green Version]
- Igreja, S.; Chahal, H.S.; Akker, S.A.; Gueorguiev, M.; Popovic, V.; Damjanovic, S.; Burman, P.; Wass, J.A.; Quinton, R.; Grossman, A.B.; et al. Assessment of p27 (cyclin-dependent kinase inhibitor 1B) and aryl hydrocarbon receptor-interacting protein (AIP) genes in multiple endocrine neoplasia (MEN1) syndrome patients without any detectable MEN1 gene mutations. Clin. Endocrinol. 2009, 70, 259–264. [Google Scholar] [CrossRef]
- Occhi, G.; Regazzo, D.; Trivellin, G.; Boaretto, F.; Citato, D.; Bobisse, S.; Ferasin, S.; Cetani, F.; Pardi, E.; Korbonits, M.; et al. A Novel Mutation in the Upstream Open Reading Frame of the CDKN1B Gene Causes a MEN4 Phenotype. PLoS Genet. 2013. [Google Scholar] [CrossRef]
- Sambugaro, S.; Di Ruvo, M.; Ambrosio, M.R.; Pellegata, N.S.; Bellio, M.; Guerra, A.; Buratto, M.; Foschini, M.P.; Tagliati, F.; Uberti, E.; et al. Early onset acromegaly associated with a novel deletion in CDKN1B 5′UTR region. Endocrine 2015, 49, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, A.; Rossing, M.; Hermann, P.; Ejersted, C.; Thakker, R.V.; Frost, M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J. Clin. Endocrinol. Metab. 2019, 104, 3637–3646. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, B.; Odou, M.F.; Demonchy, J.; Cardot-Bauters, C.; Vantyghem, M.C. Multiple Endocrine Neoplasia Type 4: Novel CDNK1B variant and immune anomalies. Ann. Endocrinol. 2020, 81, 124–125. [Google Scholar] [CrossRef]
- Chasseloup, F.; Pankratz, N.; Lane, J.; Faucz, F.R.; Keil, M.F.; Chittiboina, P.; Kay, D.M.; Tayeb, T.H.; Stratakis, C.A.; Mills, J.L.; et al. Germline CDKN1B Loss-of-Function Variants Cause Pediatric Cushing’s Disease With or Without an MEN4 Phenotype. J. Clin. Endocrinol. Metab. 2020, 105, 1983–2005. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Mateo, C.M.; Marx, S.J. Rare Germline Mutations in Cyclin-Dependent Kinase Inhibitor Genes in Multiple Endocrine Neoplasia Type 1 and Related States. J. Clin. Endocrinol. Metab. 2009, 94, 1826–1834. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.J.O.; Christie, P.T.; Pearce, S.H.S.; Turnpenny, P.D.; Thakker, R.V. Diagnostic challenges due to phenocopies: Lessons from Multiple Endocrine Neoplasia type1 (MEN1). Hum. Mutat. 2010. [Google Scholar] [CrossRef]
- Backman, S.; Bajic, D.; Crona, J.; Hellman, P.; Skogseid, B.; Stålberg, P. Whole genome sequencing of apparently mutation-negative MEN1 patients. Eur. J. Endocrinol. 2020, 182, 35–45. [Google Scholar] [CrossRef]
- Salenave, S.; Boyce, A.M.; Collins, M.T.; Chanson, P. Acromegaly and mccune-albright syndrome. J. Clin. Endocrinol. Metab. 2014, 99, 1955–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008. [Google Scholar] [CrossRef] [Green Version]
- Nerlich, A.; Peschel, O.; Löhrs, U.; Parsche, F.; Betz, P. Juvenile gigantism plus polyostotic fibrous dysplasia in the Tegernsee giant. Lancet 1991, 338, 886–887. [Google Scholar] [CrossRef]
- Aflorei, E.D.; Korbonits, M. Epidemiology and etiopathogenesis of pituitary adenomas. J. Neurooncol. 2014, 117, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Spada, A.; Vallar, L. G-protein oncogenes in acromegaly. Horm. Res. 1992, 117, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, L.S.; Shenker, A.; Friedman, E.; Spiegel, A.M.; Gejman, P.V.; Merino, M.J. Activating mutations of the stimulatory g protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- Shenker, A.; Weinstein, L.S.; Sweet, D.E.; Spiegel, A.M. An activating Gs alpha mutation is present in fibrous dysplasia of bone in the McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 1994, 79, 750–755. [Google Scholar] [PubMed]
- Schwindinger, W.F.; Francomano, C.A.; Levine, M.A. Identification of a mutation in the gene encoding the α subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc. Natl. Acad. Sci. USA 1992, 89, 5152–5156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanet, P.; Philibert, P.; Fina, F.; Cuny, T.; Roche, C.; Ouafik, L.; Paris, F.; Reynaud, R.; Barlier, A. Using Digital Droplet Polymerase Chain Reaction to Detect the Mosaic GNAS Mutations in Whole Blood DNA or Circulating Cell-Free DNA in Fibrous Dysplasia and McCune-Albright Syndrome. J. Pediatr. 2019, 205, 281–285.e4. [Google Scholar] [CrossRef]
- Wong, S.C.; Zacharin, M. Long-term health outcomes of adults with McCune-Albright syndrome. Clin. Endocrinol. 2017, 87, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Bertherat, J.; Horvath, A.; Groussin, L.; Grabar, S.; Boikos, S.; Cazabat, L.; Libe, R.; Rene-Corail, F.; Stergiopoulos, S.; Bourdeau, I.; et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J. Clin. Endocrinol. Metab. 2019, 94, 2085–2091. [Google Scholar] [CrossRef] [Green Version]
- Cuny, T.; Mac, T.T.; Romanet, P.; Dufour, H.; Morange, I.; Albarel, F.; Lagarde, A.; Castinetti, F.; Graillon, T.; North, M.O.; et al. Acromegaly in Carney complex. Pituitary 2019, 22, 456–466. [Google Scholar] [CrossRef]
- Correa, R.; Salpea, P.; Stratakis, C.A. Carney complex: An update. Eur. J. Endocrinol. 2015, 173, M85–M97. [Google Scholar] [CrossRef]
- Boikos, S.A.; Stratakis, C.A. Carney complex: The first 20 years. Curr. Opin. Oncol. 2007, 19, 24–29. [Google Scholar] [CrossRef]
- Carney, A.J.; Gordon, H.; Carpenter, P.C.; Shenoy, V.B.; Go, V.L. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine 1985, 64, 270–283. [Google Scholar] [CrossRef]
- Tsay, C.J.; Stratakis, C.A.; Faucz, F.R.; London, E.; Stathopoulou, C.; Allgauer, M.; Quezado, M.; Dagradi, T.; Spencer, D.D.; Lodish, M. Harvey Cushing Treated the First Known Patient With Carney Complex. J. Endocr. Soc. 2017, 1, 1312–1321. [Google Scholar] [CrossRef] [Green Version]
- Espiard, S.; Bertherat, J. Carney complex. Front. Horm. Res. 2013, 41, 50–62. [Google Scholar] [PubMed]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Salpea, P.; Horvath, A.; London, E.; Faucz, F.R.; Vetro, A.; Levy, I.; Gourgari, E.; Dauber, A.; Holm, I.A.; Morisson, P.J.; et al. Deletions of the PRKAR1A locus at 17q24.2–q24.3 in Carney complex: Genotype-phenotype correlations and implications for genetic testing. J. Clin. Endocrinol. Metab. 2014, 99, E183–E188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlino, A.; Vetro, A.; Garavelli, L.; Ciccone, R.; London, E.; Stratakis, C.A.; Zuffardi, O. PRKACB and Carney complex. N. Engl. J. Med. 2014, 370, 1065–1067. [Google Scholar] [CrossRef]
- Forlino, A.; Vetro, A.; Garavelli, L.; Ciccone, R.; London, E.; Stratakis, C.A.; Zuffardi, O. Genetic Diagnosis in Whole Genome Sequencing. N. Engl. J. Med. 2014, 1067–1069. [Google Scholar]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Stelmachowska-Banaś, M.; Zgliczyński, W.; Tutka, P.; Carney, A.J.; Korbonits, M. Fatal carney complex in siblings due to de novo large gene deletion. J. Clin. Endocrinol. Metab. 2017, 102, 3924–3927. [Google Scholar] [CrossRef]
- Matyakhina, L.; Bei, T.A.; McWhinney, S.R.; Pasini, B.; Cameron, S.; Gunawan, B.; Stergiopoulos, S.G.; Bikos, S.; Muchow, M.; Dutra, A.; et al. Genetics of carney triad: Recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2938–2943. [Google Scholar] [CrossRef] [Green Version]
- Stratakis, C.A.; Carney, A.J.; Lin, J.P.; Papanicolaou, D.A.; Karl, M.; Kastner, D.L.; Pras, E.; Chrousos, G.P. Carney complex, a familial multiple neoplasia and lentiginosis syndrome: Analysis of 11 kindreds and linkage to the short arm of chromosome. J. Clin. Investig. 1996, 97, 699–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivesen, K. Acromegaly Associated with Phæochromocytoma. Acta Med. Scand. 1952, 142, 1–5. [Google Scholar] [CrossRef] [PubMed]
- van Nederveen, F.H.; Gaal, J.; Favier, J.; Korpershoek, E.; Oldenburg, R.A.; de Bruyn, E.M.C.A.; Sleddens, H.F.B.M.; Derkx, P.; Riviere, J.; Dannenberg, H.; et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: A retrospective and prospective analysis. Lancet Oncol. 2009, 10, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Xekouki, P.; Pacak, K.; Almeida, M.; Wassif, C.A.; Rustin, P.; Nesterova, M.; de la Luz Sierra, M.; Matro, J.; Ball, E.; Azevedo, M.; et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: A new association for SDH? J. Clin. Endocrinol. Metab. 2012. [Google Scholar] [CrossRef]
- O’Toole, S.M.; Dénes, J.; Robledo, M.; Stratakis, C.A.; Korbonits, M. The association of pituitary adenomas and phaeochromocytomas or paragangliomas. Endocr. Relat. Cancer 2015, 22, T105–T122. [Google Scholar] [CrossRef]
- Mougel, G.; Lagarde, A.; Albarel, F.; Essamet, W.; Luigi, P.; Mouly, C.; Vialon, M.; Cuny, T.; Castinetti, F.; Saveanu, A.; et al. Germinal defects of SDHx genes in patients with isolated pituitary adenoma. Eur. J. Endocrinol. 2020, 183, 369–379. [Google Scholar] [CrossRef]
- Xekouki, P.; Brennand, A.; Whitelaw, B.; Pacak, K.; Stratakis, C.A. The 3PAs: An Update on the Association of Pheochromocytomas, Paragangliomas, and Pituitary Tumors. Horm. Metab. Res. 2019, 51, 419. [Google Scholar] [CrossRef] [Green Version]
- Dénes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell. 2015, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.J.C.M.M.; Sacre, N.; van der Lely, A.J.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr. Relat. Cancer 2018, 25, L37–L42. [Google Scholar] [CrossRef] [Green Version]
- Roszko, K.L.; Blouch, E.; Blake, M.; Powers, J.F.; Tischler, A.S.; Hodin, R.; Sadow, P.; Lawson, E.A. Case Report of a Prolactinoma in a Patient With a Novel MAX Mutation and Bilateral Pheochromocytomas. J. Endocr. Soc. 2017, 1, 1401–1407. [Google Scholar] [CrossRef] [Green Version]
- Romero, O.A.; Torres-Diz, M.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Saez, C.; Iwaka, R.; Villanueva, A.; Montuenga, L.M.; et al. MAX inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with BRG. Cancer Discov. 2014, 4, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Carroll, P.A.; Freie, B.W.; Mathsyaraja, H.; Eisenman, R.N. The MYC transcription factor network: Balancing metabolism, proliferation and oncogenesis. Front. Med. 2018, 12, 412–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comino-Méndez, I.; Gracia-Aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-Garcia, L.J.; Leton, R.; Honrado, E.; Ramos-Medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Gen. 2011, 43, 663. [Google Scholar] [CrossRef]
- Stütz, B.; Korbonits, M.; Kothbauer, K.; Müller, W.; Fischli, S. Identification of a TMEM127 variant in a patient with paraganglioma and acromegaly. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020, 20-0119. [Google Scholar] [CrossRef]
- Guerrero-Pérez, F.; Fajardo, C.; Torres Vela, E.; Gimenez-Palop, O.; Gil, A.L.; Martin, T.; Gonzalez, N.; Diez, J.J.; Iglesias, P.; Robledo, M.; et al. 3P association (3PAs): Pituitary adenoma and pheochromocytoma/paraganglioma. A heterogeneous clinical syndrome associated with different gene mutations. Eur. J. Intern Med. 2019, 69, 14–19. [Google Scholar] [CrossRef]
- Vieira Neto, L.; Taboada, G.F.; Corrêa, L.L.; Polo, J.; Nascimento, A.F.; Chimelli, L.; Rumilla, K.; Gadelha, M.R. Acromegaly secondary to growth hormone-releasing hormone secreted by an incidentally discovered pheochromocytoma. Endocr. Pathol. 2017, 18, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, A.A.; Amirbaigloo, A.; Dezfooli, A.A.; Sadat, N.; Ghazi, S.; Pourafkari, M.; Tigari, F.; Dhall, D.; Bannykh, S.; Melmed, S.; et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine 2013, 43, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Seabrook, A.J.; Harris, J.E.; Velosa, S.B.; Kim, E.; McInerney-Leo, A.M.; Dwight, T.; Hockings, J.I.; Hockings, N.G.; Kirk, J.; Leo, P.J.; et al. Multiple Endocrine Tumors Associated with Germline MAX Mutations: Multiple Endocrine Neoplasia Type 5? J. Clin. Endocrinol. Metab. 2020. [Google Scholar] [CrossRef]
- Josefson, J.L.; Listernick, R.; Charrow, J.; Habiby, R.L. Growth Hormone Excess in Children with Optic Pathway Tumors Is a Transient Phenomenon. Horm. Res. Paediatr. 2016, 86, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, C.; Bottaro, G. Endocrine implications of neurofibromatosis 1 in childhood. Horm. Res. Paediatr. 2015, 83, 232–241. [Google Scholar] [CrossRef]
- Hannah-Shmouni, F.; Stratakis, C.A. Growth hormone excess in neurofibromatosis. Genet. Med. 2019, 21, 1254–1255. [Google Scholar] [CrossRef]
- Hozumi, K.; Fukuoka, H.; Odake, Y.; Takeuchi, T.; Uehara, T.; Sato, T.; Inoshita, N.; Yoshida, K.; Matsumoto, R.; Bando, H.; et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type. Endocr. J. 2019, 66, 853–857. [Google Scholar] [CrossRef] [Green Version]
- Checa Garrido, A.; del Pozo Picó, C. Acromegaly and type 1 neurofibromatosis. Is association of both conditions due to chance? Endocrinol. Nutr. 2013, 60, 144–145. [Google Scholar] [CrossRef]
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Bruce, B.; Maria, B.L. Neurofibromatosis type 1 revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, D.; Pezzani, L.; Tadini, G.; Menni, F.; Esposito, S. A multidisciplinary approach in neurofibromatosis. Lancet Neurol. 2015, 14, 29–30. [Google Scholar] [CrossRef]
- Faucz, F.R.; Horvath, A.D.; Azevedo, M.F.; Levy, I.; Bak, B.; Wang, Y.; Xekouki, P.; Szarek, E.; Gourgari, E.; Manning, A.D.; et al. Is IGSF1 involved in human pituitary tumor formation? Endocr. Relat. Cancer 2015, 22, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, W.H.; Perrin, J.C.S.; Halac, E.; Gala, R.R.; England, B.G. Acromegalic gigantism and tuberous sclerosis. J. Pediatr. 1978, 93, 478–480. [Google Scholar] [CrossRef]
- Tigas, S.; Carroll, P.V.; Jones, R.; Bingham, E.; Russell-Jones, D.; Powell, M.; Scobie, I.N. Simultaneous Cushing’s disease and tuberous sclerosis; a potential role for TSC in pituitary ontogeny. Clin. Endocrinol. 2005, 63, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Nandagopal, R.; Vortmeyer, A.; Oldfield, E.H.; Keil, M.F.; Stratakis, C.A. Cushing’s syndrome due to a pituitary corticotropinoma in a child with tuberous sclerosis: An association or a coincidence? Clin. Endocrinol. 2007, 67, 639–641. [Google Scholar] [CrossRef]
- Regazzo, D.; Gardiman, M.P.; Theodoropoulou, M.; Scaroni, C.; Occhi, G.; Ceccato, F. Silent gonadotroph pituitary neuroendocrine tumor in a patient with tuberous sclerosis complex: Evaluation of a possible molecular link. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neou, M.; Villa, C.; Armignacco, R.; Jouinot, A.; Raffin-Sanson, M.L.; Septier, A.; Letourneur, F.; Diry, S.; Diedisheim, M.; Izac, B.; et al. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell. 2020, 37, 123–134.e5. [Google Scholar] [CrossRef]
- Song, Z.J.; Reitman, Z.J.; Ma, Z.Y.; Chen, J.H.; Zhang, Q.L.; Shou, X.F.; Huang, C.X.; Wang, Y.F.; Li, S.Q.; Mao, Y.; et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res. 2016, 26, 1255–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, R.; Izawa, M.; Fukuoka, H.; Iguchi, G.; Odake, Y.; Yoshida, K.; Bando, H.; Suda, K.; Nishizawa, H.; Takahashi, M.; et al. Genetic and clinical characteristics of Japanese patients with sporadic somatotropinoma. Endocr. J. 2016, 63, 953–963176. [Google Scholar] [CrossRef] [Green Version]
- Bakhtiar, Y.; Hirano, H.; Arita, K.; Yunoue, S.; Fujio, S.; Tominaga, A.; Sakoguchi, T.; Sugiyama, K.; Kurisu, K.; Yasufuku-Takano, J.; et al. Relationship between cytokeratin staining patterns and clinico-pathological features in somatotropinomae. Eur. J. Endocrinol. 2010, 163, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.; Buslei, R.; Theodoropoulou, M.; Stalla, G.K.; Buchfelder, M.; Schöfl, C. Molecular and functional properties of densely and sparsely granulated GH-producing pituitary adenomas. Eur. J. Endocrinol. 2013, 169, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, B.E.; Barlier, A.; Korbonits, M.; Grossman, A.B.; Jacquet, P.; Enjalbert, A.; Bonthron, D.T. Imprinting of the Gsα gene GNAS1 in the pathogenesis of acromegaly. J. Clin. Investig. 2001. [Google Scholar] [CrossRef] [Green Version]
- Gadelha, M.R.; Trivellin, G.; Hernández-Ramírez, L.C.; Korbonits, M. Genetics of Pituitary Adenomas. Endocr. Tumor Syndr. Genet. Front Horm. Res. Basel. Karger. 2013, 41, 111–140. [Google Scholar]
- Regazzo, D.; Losa, M.; Albiger, N.M.; Terreni, M.R.; Vazza, G.; Ceccato, F.; Emanuelli, E.; Denaro, L.; Scaroni, C.; Occhi, G. The GIP/GIPR axis is functionally linked to GH-secretion increase in a significant proportion of gsp-somatotropinomas. Eur. J. Endocrinol. 2017, 176, 543–553. [Google Scholar] [CrossRef]
- Scaroni, C.; Albiger, N.; Daniele, A.; Dassie, F.; Romualdi, C.; Vazza, G.; Regazzo, D.; Ferrau, F.; Barresi, V.; Maffeis, V.; et al. Paradoxical GH Increase during OGTT Is Associated with First-Generation Somatostatin Analog Responsiveness in Acromegaly. J. Clin. Endocrinol. Metab. 2018, 104, 856–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilezikjian, L.M.; Erlichman, J.; Fleischer, N.; Vale, W.W. Differential activation of type I and type II 3′,5′-cyclic adenosine monophosphate-dependent protein kinases by growth hormone releasing factor. Mol. Endocrinol. 1987, 1, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, M.J.; Kim, H.Y.; Kim, S.G.; Park, J.; Choi, D.S.; Hwang, J.I.; Seong, J.Y. Tyr1 and Ile7 of glucose-dependent insulinotropic polypeptide (GIP) confer differential ligand selectivity toward GIP and glucagon-like peptide-1 receptors. Mol. Cells 2010, 30, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Hage, M.; Chaligné, R.; Viengchareun, S.; Villa, C.; Salenave, S.; Bouligand, J.; Letouze, E.; Tosca, L.; Rouguette, A.; Tachdjian, G.; et al. Hypermethylator Phenotype and Ectopic GIP Receptor in GNAS Mutation-Negative Somatotropinomas. J. Clin. Endocrinol. Metab. 2019, 104, 1777–1787. [Google Scholar] [CrossRef]
- Schilbach, K.; Gar, C.; Lechner, A.; Nociolay, S.S.; Schwerdt, L.; Haenelt, M.; Dal, J.; Jorgensen, J.O.L.; Stormann, S.; Schopohl, J.; et al. Determinants of the growth hormone nadir during oral glucose tolerance test in adults. Eur. J. Endocrinol. 2019, 181, 55–67. [Google Scholar] [CrossRef]
- Ronchi, C.L.; Peverelli, E.; Herterich, S.; Weigand, I.; Mantovani, G.; Schwarzmayr, T.; Sbiera, S.; Allilio, B.; Honegger, J.; Appenzeller, S.; et al. Landscape of somatic mutations in sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2016, 174, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Välimäki, N.; Demir, H.; Pitkänen, E.; Kaasinen, E.; Karppinen, A.; Kivipelto, L.; Schalin-Jäntti, C.; Aaltonen, L.A.; Karhu, A. Whole-genome sequencing of growth hormone (GH)-secreting pituitary adenomas. J. Clin. Endocrinol. Metab. 2015, 100, 3918–3927. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.P.Y.; Kelsey, M.M.; Sabbaghian, N.; Park, S.H.; Deal, C.L.D.; Esbenshade, A.J.; Ploner, O.; Peet, A.; Traunecker, H.; Ahmed, Y.H.E.; et al. Clinical Outcomes and Complications of Pituitary Blastoma. J. Clin. Endocrinol. Metab. 2020, 106, 351–363. [Google Scholar] [CrossRef]
- Nadhamuni, V.S.; Korbonits, M. Novel insights into pituitary tumorigenesis: Genetic and epigenetic mechanisms. Endocr. Rev. 2020. [Google Scholar] [CrossRef] [Green Version]
- Marques, P.; Grossman, A.B.; Korbonits, M. The Tumour Microenvironment of Pituitary Neuroendocrine Tumours. Front. Neuroendocrinol. 2020, 58, 100852. [Google Scholar] [CrossRef]
- Marques, P.; Barry, S.; Carlsen, E.; Carlsen, E.; Collier, D.; Ronaldson, A.; Awad, S.; Dorward, N.; Grieve, J.; Mendoza, N.; et al. Pituitary tumour fibroblast-derived cytokines influence tumour aggressiveness. Endocr. Relat. Cancer 2019, 26, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Caimari, F.; Hernández-Ramírez, L.C.; Dang, M.N.; Gabrovska, P.; Iacovazzo, D.; Stals, K.; Ellard, S.; Korbonits, M. International FIPA consortium. Risk category system to identify pituitary adenoma patients with AIP mutations. J. Med. Genet. 2018, 55, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
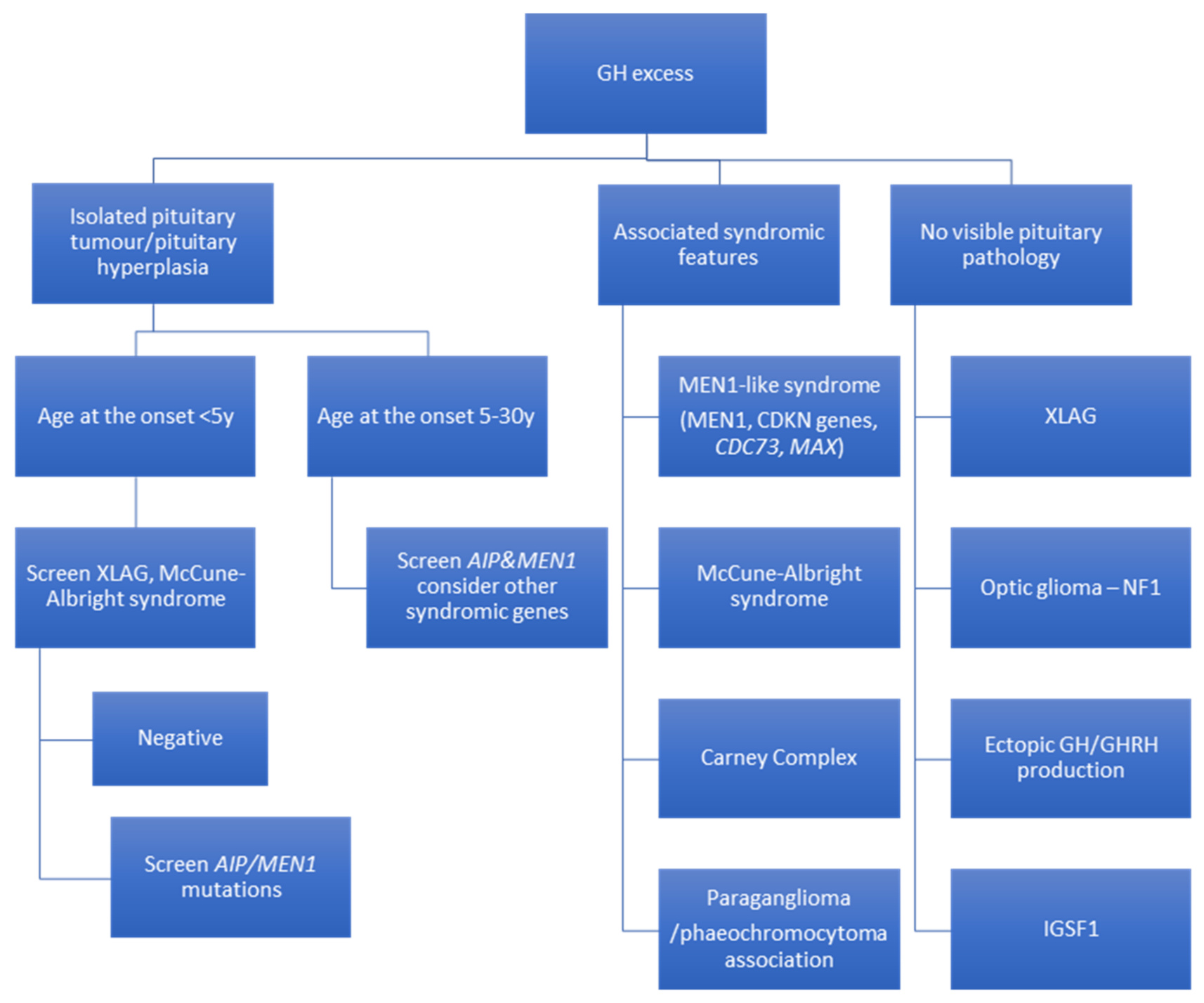

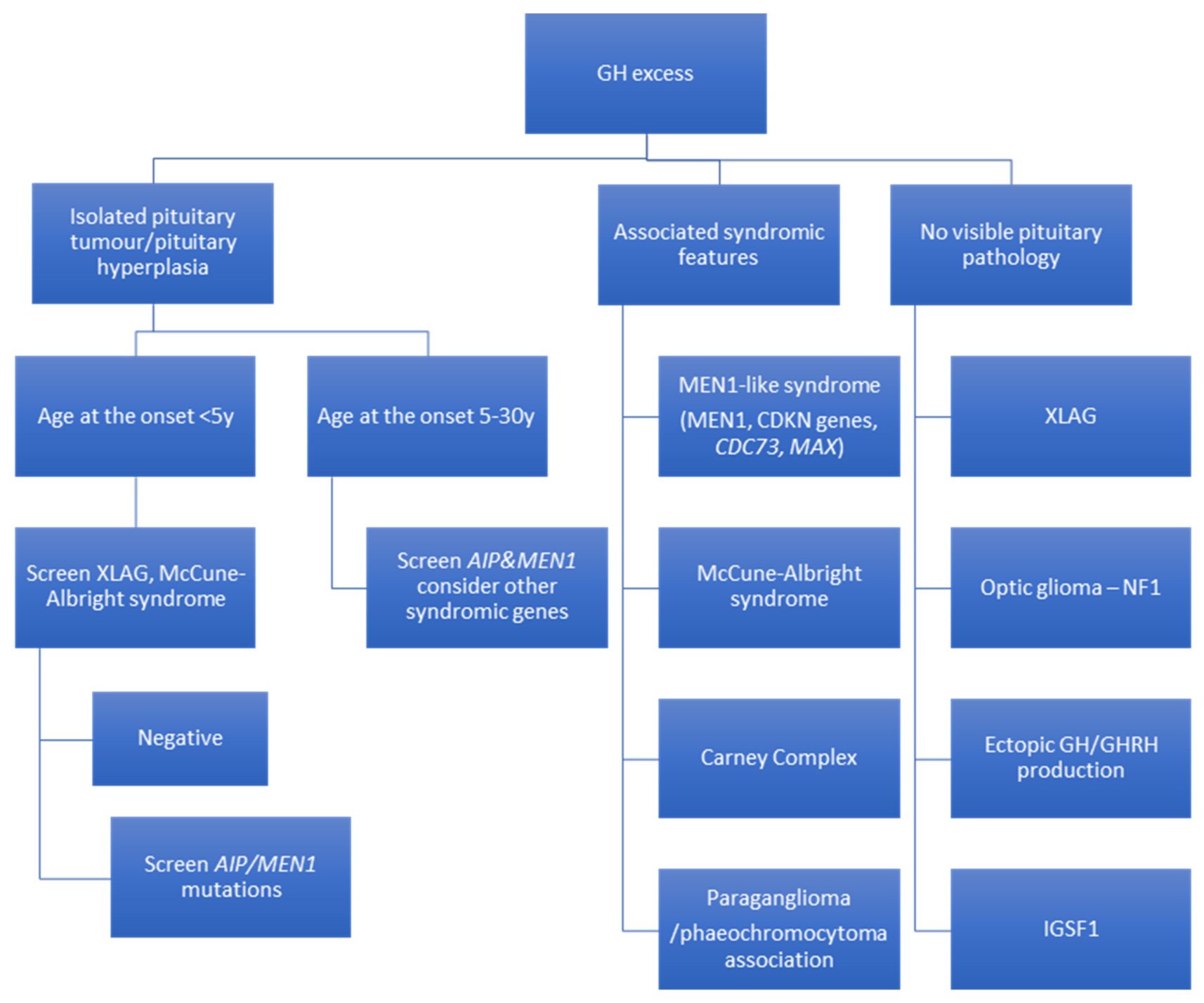{kind=link}
| Disease | Gene Mutation/Genetic Alteration | Gene Location | Prevalence in Pituitary Tumours | Prevalence in Acromegaly (%) | Phenotype | Mean Age of Diagnosis of GH Excess | Histopathology |
|---|---|---|---|---|---|---|---|
| FIPA/AIP | AIP | 11q13.3 | 3.6% | 50% in homogeneous FIPA 4% in sporadic acromegaly 29% in gigantism patients | Isolated pituitary tumour | 2nd decade of life (<30 years), male predominance, reduced SSTR 2 expression | More often sparsely granulated variant |
| FIPA/X-linked acrogigantism | GPR101 | Xq26.3 | 1.6% | 0–4.4% in acromegaly 10% of gigantism patients | Isolated pituitary tumour | first years of life (<5 years) female predominance, pituitary hyperplasia or tumour, males can be mosaic or familial | Often somatotroph h/lactotroph pituitary hyperplasia in 25% of cases |
| Multiple Endocrine Neoplasia type 1 | MEN1 | 11q13.1 | 0.6–2.6% | 1.2% in acromegaly 1% of gigantism patients | Hyperparathyroidism, pituitary tumour, pancreatic neuroendocrine tumours | 4th decade of life female predominance | Multiple PAs and more often plurihormonal profile. More often pituitary hyperplasia. In some part of patients, poorly-differentiated PIT1- lineage tumours |
| Multiple Endocrine Neoplasia type 4 | CDKN1B | 12p13.1 | rare | rare | Hyperparathyroidism, pituitary tumour, pancreatic neuroendocrine tumours | Single cases | More often pituitary hyperplasia |
| McCune–Albright Syndrome | Mosaic GNAS mutation | 20q13.3 | Only acromegaly/gigantism (20% of patients) | 5% of gigantism patients | Classic triad: fibrosus dysplasia, cafe- au-lait macules, precocious puberty | 2nd decade of life male predominance, pituitary hyperplasia, prolactin cosecretion | More often pituitary hyperplasia |
| Carney Complex | PRKAR1A | 17q22-24 | Only acromegaly/gigantism (12% but 75% asymptomatic elevation of GH and IGF-1 | 1% among gigantism patients | Acromegaly, cardiac and cutaneous myxomas, PPNAD, lentiginosis | 3rd decade of life no gender predominance, hyperplasia (majority) or tumour | somatotroph h/lactotroph pituitary hyperplasia |
| CNC2 locus | 2p16 | ||||||
| Pituitary adenoma and PPGL association | SDHx VHL MEN1 RET | SDHA 5p15.33 SDHB 1p36.13 SDHC 1q23.3 SDHD 11q23.1 | rare | rare | Association between PPGL and pituitary tumour | Single cases | intracytoplasmic vacuoles |
| MAX | 14q23.3 | ||||||
| Neurofibromatosis type 1 | NF1 | 17q11.2 | Only acromegaly/gigantism- around 10% in patients with NF1 and optic glioma | rare | Neurofibromas, cafe au-lait macules, freckling, Lisch nodules, optic glioma | No visible pituitary pathology | - |
| Deficiency of the X-link immunoglobulin superfamily member 1 | IGSF1 | Xq26.1 | Only GH excess features | Not estimated | acromegalic facial features organomegaly in adulthood | No visible pituitary pathology | - |
| Sporadic somatotropinomas | Somatic GNAS mutation | 20q13.3 | Only acromegaly | 40% | Isolated pituitary tumour | smaller size, good response to medical treatment with somatostatin analogues | no association has been observed between GNAS mutation and granulation pattern |
| cAMP Pathway | Calcium Signalling | ATP Signalling |
|---|---|---|
| GNAS PRKAA2 ADRBK2 ATP6V0A1 CCR10 CHRM3 OR51B4 GNAQ | CACNA1H CAPN1 DMD GRIN2B JPH2 MAN1A1 PCDH11X PROCA1 SLIT2 SPTA1 TESC C2CD3 RYR1 SSR3 WIPI1 | SUPV3L1 ATPAF2 ATAD2B DICER1 AOX1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogusławska, A.; Korbonits, M. Genetics of Acromegaly and Gigantism. J. Clin. Med. 2021, 10, 1377. https://doi.org/10.3390/jcm10071377
Bogusławska A, Korbonits M. Genetics of Acromegaly and Gigantism. Journal of Clinical Medicine. 2021; 10(7):1377. https://doi.org/10.3390/jcm10071377
Chicago/Turabian StyleBogusławska, Anna, and Márta Korbonits. 2021. "Genetics of Acromegaly and Gigantism" Journal of Clinical Medicine 10, no. 7: 1377. https://doi.org/10.3390/jcm10071377
APA StyleBogusławska, A., & Korbonits, M. (2021). Genetics of Acromegaly and Gigantism. Journal of Clinical Medicine, 10(7), 1377. https://doi.org/10.3390/jcm10071377