
Variable Expressivity and Allelic Heterogeneity in Type 2 Familial Partial Lipodystrophy: The p.(Thr528Met) LMNA Variant

 , ,
, ,  ,
,  ,
, 
Abstract
:1. Introduction
2. Experimental Section
2.1. Subjects, Analysis and Interpretation of Variants
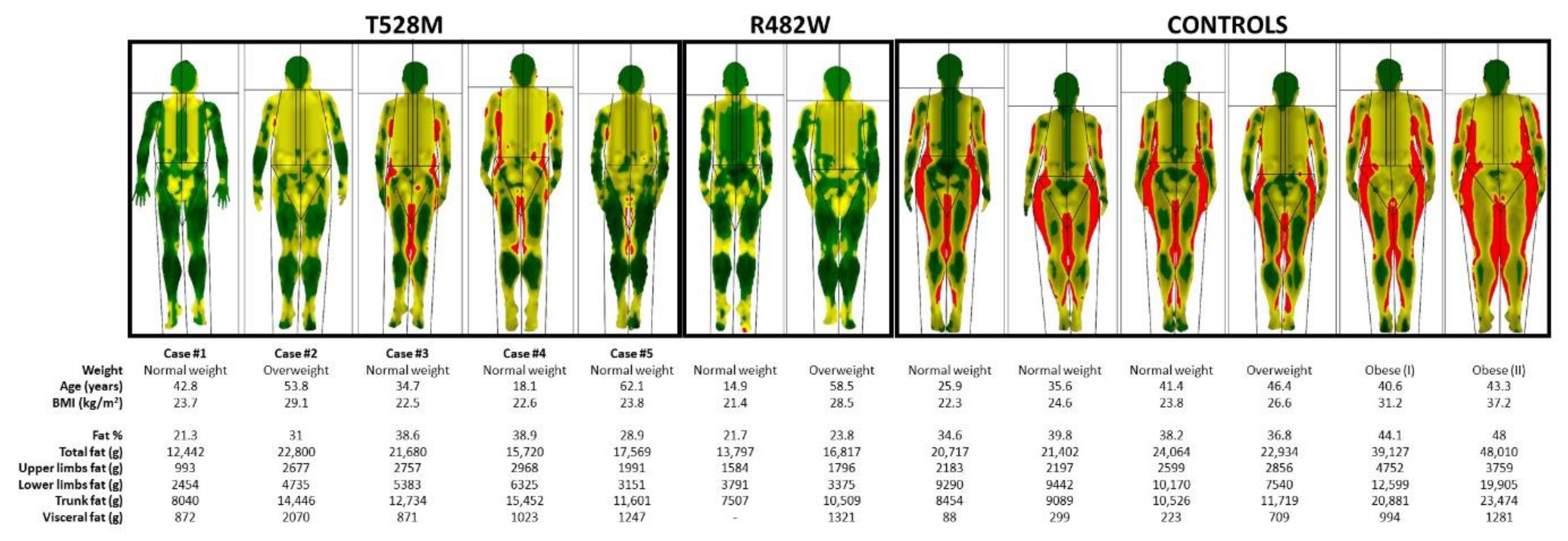
2.2. Body Composition Studies
2.3. Biochemical Analyses
2.4. Adipose Tissue Biopsies and Cell Culture
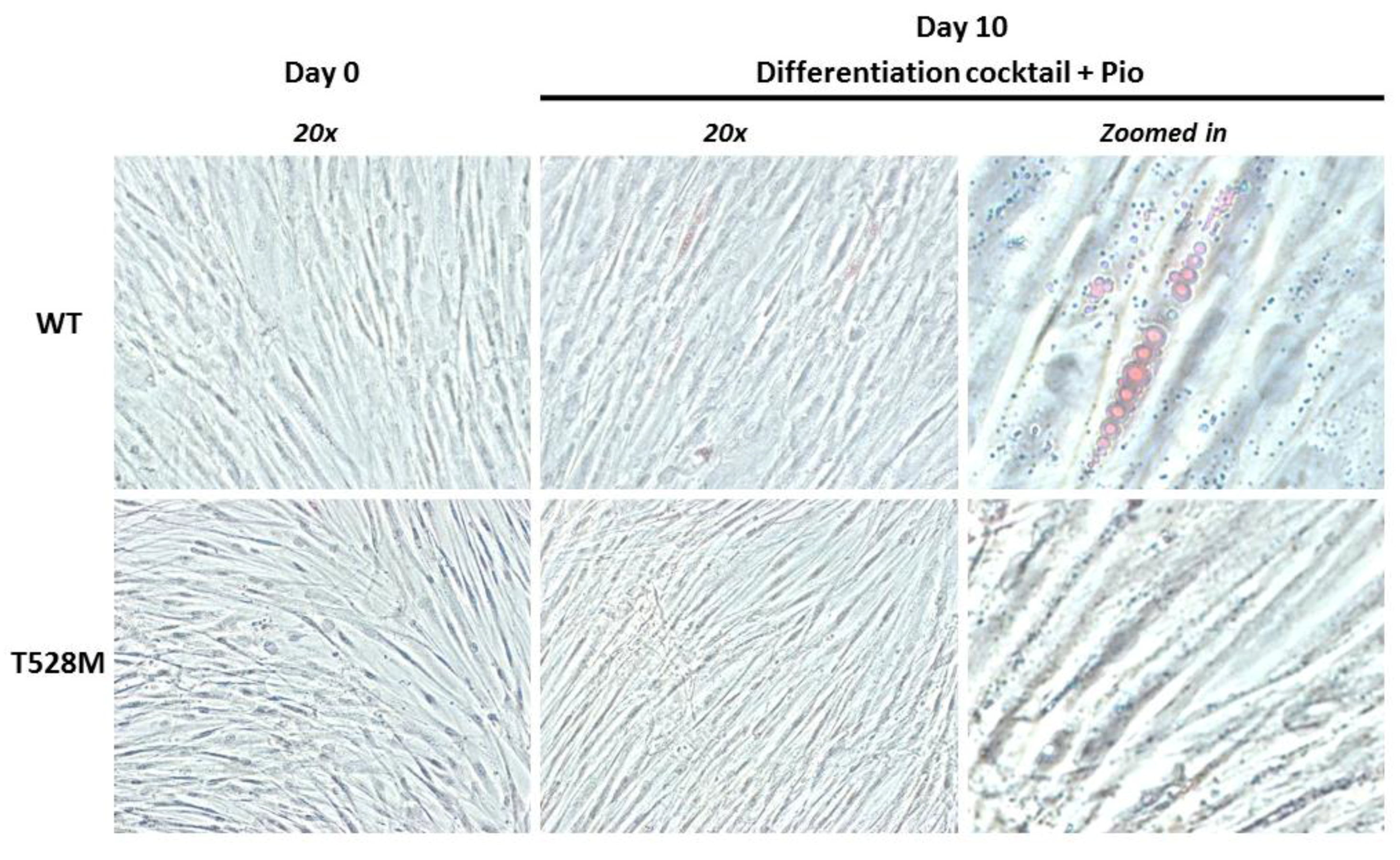
2.5. Adipocyte Differentiation Procedure
2.6. Phase Contrast Microscopy
2.7. RNA Extraction and Retrotranscription
2.8. Real-Time PCR
2.9. Immunofluorescence
2.10. Statistical Analysis
3. Results
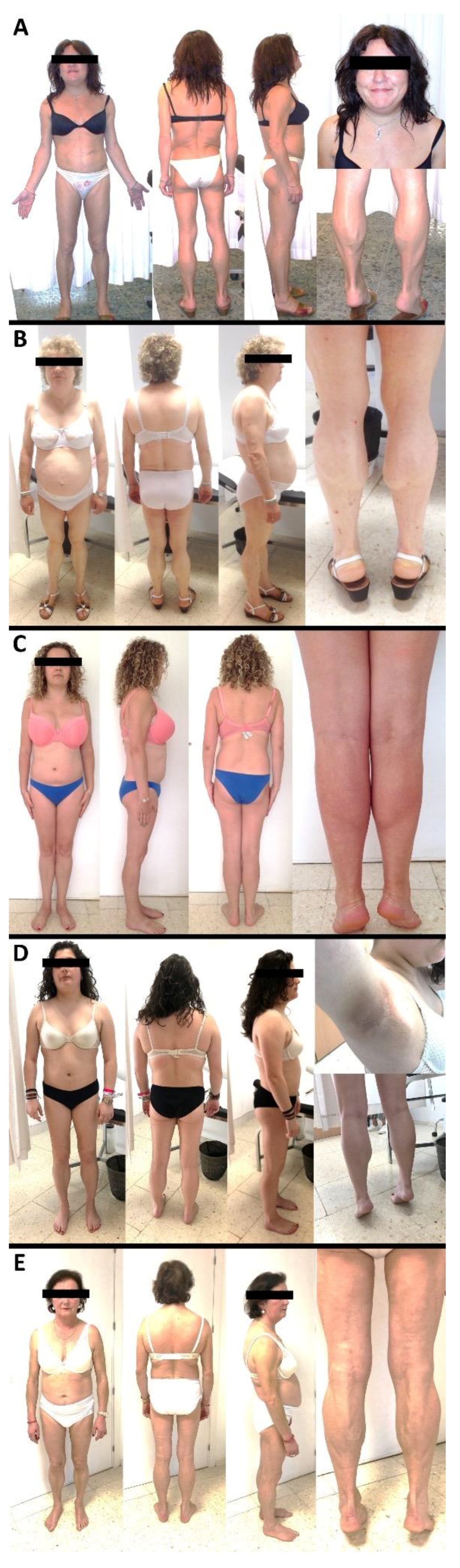
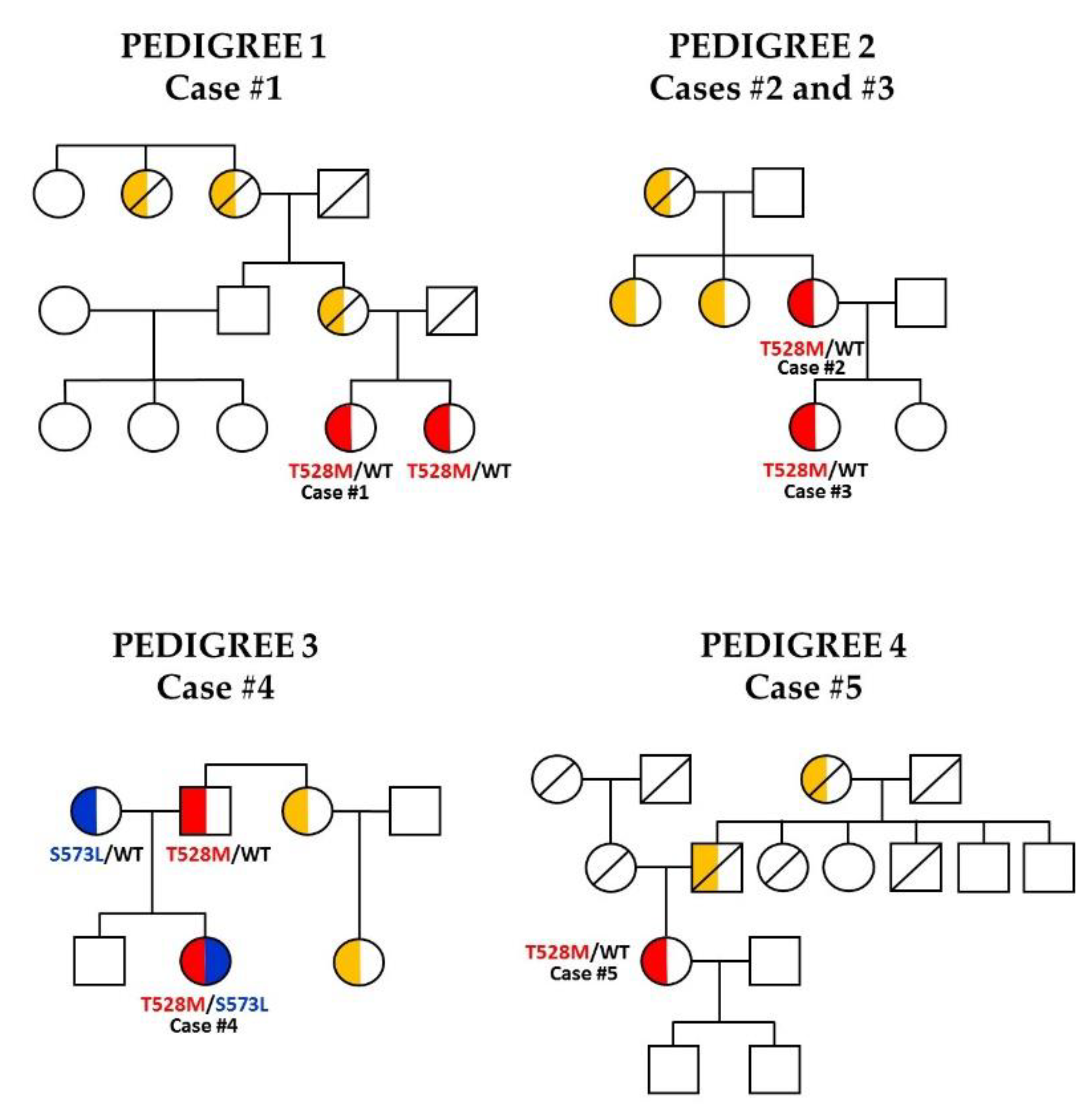
3.1. Case Reports
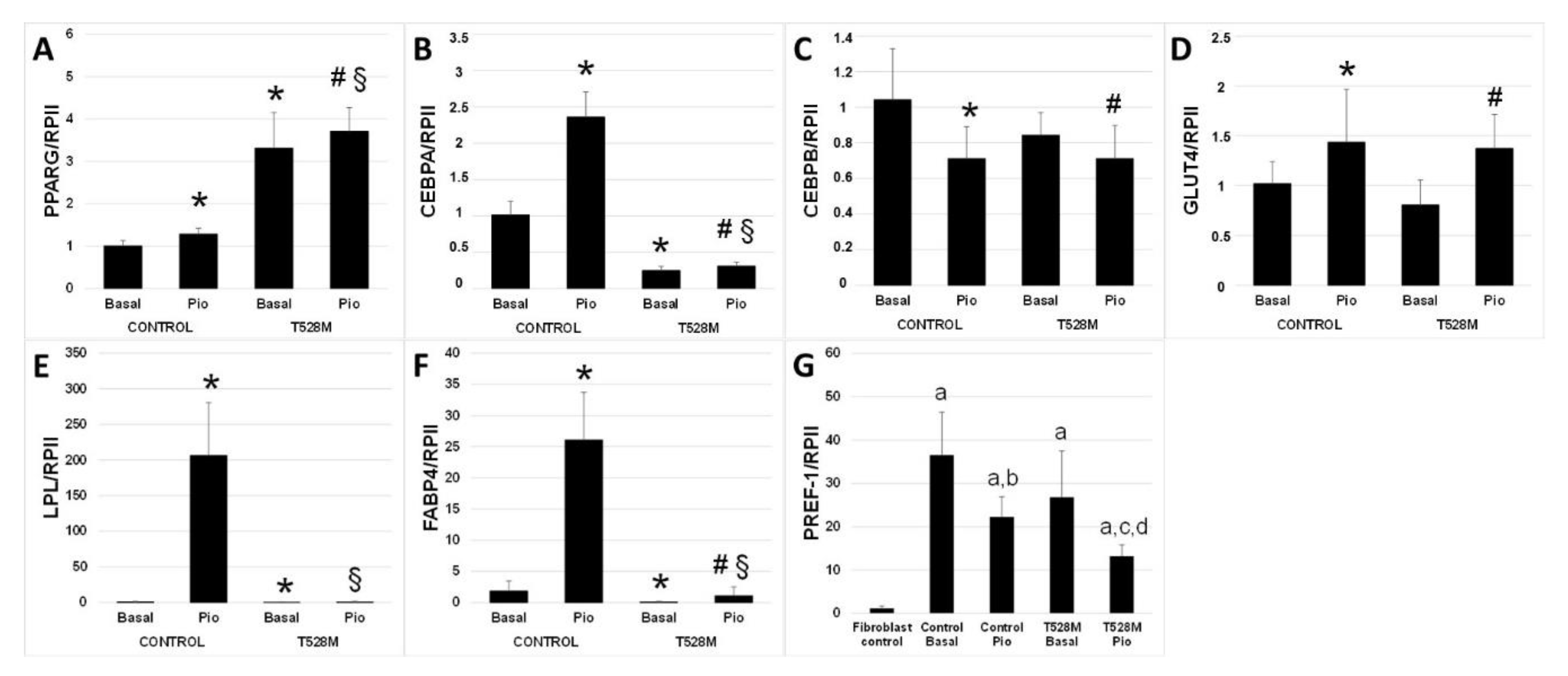
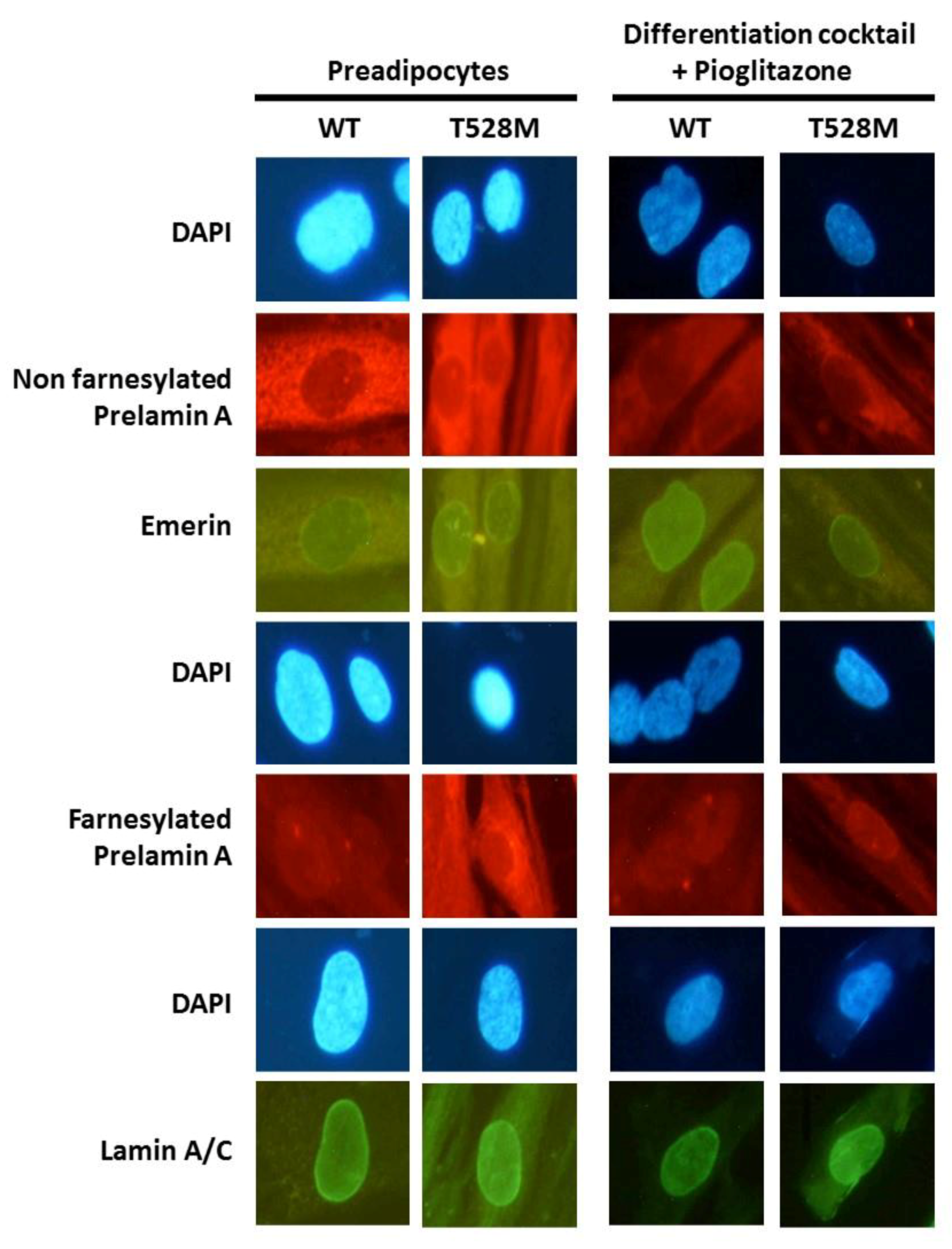
3.2. Cellular and Gene Expression Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savage, D.B.; Soos, M.A.; Powlson, A.; O’rahilly, S.; McFarlane, I.; Halsall, D.J.; Barroso, I.; Thomas, E.L.; Bell, J.D.; Scobie, I.; et al. Familial partial lipodystrophy associated with compound heterozygosity for novel mutations in the LMNA gene. Diabetologia 2004, 47, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, V.L.; Broers, J.L.; van Steensel, M.A.; Zinn-Justin, S.; Ramaekers, F.C.; Steijlen, P.M.; Kamps, M.; Kuijpers, H.J.; Merckx, D.; Smeets, H.J.; et al. Compound heterozygosity for mutations in LMNA causes a progeria syndrome without prelamin A accumulation. Hum. Mol. Genet. 2006, 15, 2509–2522. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Mazess, R.B.; Barden, H.S.; Bisek, J.P.; Hanson, J. Dual-energy x-ray absorptiometry for total-body and regional bone-mineral and soft-tissue composition. Am. J. Clin. Nutr. 1990, 51, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Vilar, D.; Loidi, L.; Dominguez, F.; Cabezas-Cerrato, J. Phenotypic gender differences in subjects with familial partial lipodystrophy (Dunnigan variety) due to a nuclear lamin A/C R482W mutation. Horm. Metab. Res. 2003, 35, 29–35. [Google Scholar] [CrossRef]
- Parpal, S.; Karlsson, M.; Thorn, H.; Stralfors, P. Cholesterol depletion disrupts caveolae and insulin receptor signaling for metabolic control via insulin receptor substrate-1, but not for mitogen-activated protein kinase control. J. Biol. Chem. 2001, 276, 9670–9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Iglesias, S.; Unruh-Pinheiro, A.; Guillin-Amarelle, C.; Gonzalez-Mendez, B.; Ruiz-Riquelme, A.; Rodriguez-Canete, B.L.; Rodriguez-Garcia, S.; Guillen-Navarro, E.; Domingo-Jimenez, R.; Araujo-Vilar, D. Skipped BSCL2 Transcript in Celia’s Encephalopathy (PELD): New Insights on Fatty Acids Involvement, Senescence and Adipogenesis. PLoS ONE 2016, 11, e0158874. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Dominici, S.; Fiori, V.; Magnani, M.; Schena, E.; Capanni, C.; Camozzi, D.; D’Apice, M.R.; Le Dour, C.; Auclair, M.; Caron, M.; et al. Different prelamin A forms accumulate in human fibroblasts: A study in experimental models and progeria. Eur. J. Histochem. 2009, 53, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shen, H.; Zhao, Z.; Bing, Q.; Hu, J. Cardiac effects of the c.1583 C-->G LMNA mutation in two families with Emery-Dreifuss muscular dystrophy. Mol. Med. Rep. 2015, 12, 5065–5071. [Google Scholar] [CrossRef] [Green Version]
- Novelli, G.; Muchir, A.; Sangiuolo, F.; Helbling-Leclerc, A.; D’Apice, M.R.; Massart, C.; Capon, F.; Sbraccia, P.; Federici, M.; Lauro, R.; et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am. J. Hum. Genet. 2002, 71, 426–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.; Cogulu, O.; Ozkinay, F.; Onay, H.; Agarwal, A.K. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J. Clin. Endocrinol. Metab. 2005, 90, 5259–5264. [Google Scholar] [CrossRef] [Green Version]
- Kosho, T.; Takahashi, J.; Momose, T.; Nakamura, A.; Sakurai, A.; Wada, T.; Yoshida, K.; Wakui, K.; Suzuki, T.; Kasuga, K.; et al. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am. J. Med. Genet. Part A 2007, 143A, 2598–2603. [Google Scholar] [CrossRef] [PubMed]
- Masugi, J.; Tamori, Y.; Mori, H.; Koike, T.; Kasuga, M. Inhibitory effect of a proline-to-alanine substitution at codon 12 of peroxisome proliferator-activated receptor-gamma 2 on thiazolidinedione-induced adipogenesis. Biochem. Biophys. Res. Commun. 2000, 268, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Araujo-Vilar, D.; Victoria, B.; Gonzalez-Mendez, B.; Barreiro, F.; Fernandez-Rodriguez, B.; Cereijo, R.; Gallego-Escuredo, J.M.; Villarroya, F.; Paneda-Menendez, A. Histological and molecular features of lipomatous and nonlipomatous adipose tissue in familial partial lipodystrophy caused by LMNA mutations. Clin. Endocrinol. 2012, 76, 816–824. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Lattanzi, G.; Gonzalez-Mendez, B.; Costa-Freitas, A.T.; Prieto, D.; Columbaro, M.; Mattioli, E.; Victoria, B.; Martinez-Sanchez, N.; Ramazanova, A.; et al. Site-dependent differences in both prelamin A and adipogenic genes in subcutaneous adipose tissue of patients with type 2 familial partial lipodystrophy. J. Med. Genet. 2009, 46, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, A.; Briand, N.; Sorensen, A.L.; Cahyani, I.; Shah, A.; Moskaug, J.O.; Collas, P. A lipodystrophy-causing lamin A mutant alters conformation and epigenetic regulation of the anti-adipogenic MIR335 locus. J. Cell Biol. 2017, 216, 2731–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capanni, C.; Mattioli, E.; Columbaro, M.; Lucarelli, E.; Parnaik, V.K.; Novelli, G.; Wehnert, M.; Cenni, V.; Maraldi, N.M.; Squarzoni, S.; et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum. Mol. Genet. 2005, 14, 1489–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, M.; Auclair, M.; Donadille, B.; Bereziat, V.; Guerci, B.; Laville, M.; Narbonne, H.; Bodemer, C.; Lascols, O.; Capeau, J.; et al. Human lipodystrophies linked to mutations in A-type lamins and to HIV protease inhibitor therapy are both associated with prelamin A accumulation, oxidative stress and premature cellular senescence. Cell Death Differ. 2007, 14, 1759–1767. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | Probe | Probe Sequences | Amplicon Length (nt) |
|---|---|---|---|---|---|
| CEBPA | GCAAATCGTGCCTTGTCAT | CTCATGGGGGTCTGCTGTAG | 12 | CTCCTTCC | 72 |
| CEBPB | CGCTTACCTCGGCTACCA | ACGAGGAGGACGTGGAGAG | 74 | CTGCTGCC | 65 |
| FABP4 | CCTTTAAAAATACTGAGATTTCCTTCA | GGACACCCCCATCTAAGGTT | 72 | TTCCTGGC | 105 |
| GLUT4 | CTGTGCCATCCTGATGACTG | CGTAGCTCATGGCTGGAACT | 67 | TGCTGGAG | 62 |
| LPL | ATGTGGCCCGGTTTATCA | CTGTATCCCAAGAGATGGACATT | 25 | CTCCTCCA | 76 |
| PPARG | GACCTGAAACTTCAAGAGTACCAAA | TGAGGCTTATTGTAGAGCTGAGTC | 39 | CTCCACCT | 95 |
| PREF-1 | GACGGGGAGCTCTGTGATAG | CATAGAGGCCATCGTCCAG | 68 | AGGAGCAG | 94 |
| RNA polymerase II | GCATCATGAACAGCGATGAG | TCATCCATCTTGTCCACCAC | 69 | GGAGGAAG | 64 |
| Demographic Features | |||||
|---|---|---|---|---|---|
| Case #1 | Case #2 | Case #3 | Case #4 | Case #5 | |
| Age | 42.8 | 53.8 | 34.7 | 18.1 | 62.1 |
| Sex | F | F | F | F | F |
| Variants | |||||
| LMNA | c.1583C>T p.(Thr528Met) | c.1583C>T p.(Thr528Met) | c.1583C>T p.(Thr528Met) | c.1583C>T p.(Thr528Met)/c.1718C>T p.(Ser573Leu) | c.1583C>T p.(Thr528Met) |
| Other genes | - | WRN c.1495A>G p.(Arg499Gly); BLM c.813G>C p.(Lys271Asn) | WRN c.1495A>G p.(Arg499Gly); BLM c.813G>C p.(Lys271Asn) | - | - |
| Autosomal dominant inheritance | yes | yes | yes | yes | yes |
| SNP PPARGp.Pro12Ala | yes | no | no | no | no |
| Clinical features | |||||
| Acanthosis | no | yes | yes | yes | no |
| Phlebomegaly | no | yes | no | yes | yes |
| Hypermuscularity | yes | yes | no | no | yes |
| Lipomas | no | no | yes | no | no |
| Goiter | no | no | no | no | yes |
| Diabetes mellitus (DM) | yes | yes | no | no (IFG) | no (IFG) |
| Dyslipidemia | IV | IIb | IV | IV | IIb |
| Steatosis | no | yes | no | no | no |
| Arterial hypertension (AHT) | yes | yes | no | no | yes |
| Cardiovascular diseases (CVDs) | no | no | no | no | yes |
| Polycystic ovary syndrome (PCOS) and obstetric complications | no | no | no | yes | no |
| Pancreatitis | no | no | no | no | no |
| Lipodystrophy onset | Childhood | Childhood | 31 years of age | Adolescence | Adolescence |
| Family background | Mother and sister: FPLD | Daughter: FPLD; Mother: DM | Mother: FPLD | Father: hypertriglyceridemia | Mother: DM |
| Anthropometric data | |||||
| Weight (kg) | 58.5 | 73.5 | 58.2 | 61.5 | 60.8 |
| Height (cm) | 157 | 159 | 161 | 165 | 160 |
| BMI (kg/m2) | 23.7 | 29.1 | 22.5 | 22.6 | 23.8 |
| Waist (cm) | 76 | 101 | 84 | 84 | 90 |
| Hip (cm) | 87 | 100 | 87 | 94 | 89 |
| Waist-to-height ratio | 0.9 | 1 | 1 | 0.9 | 1 |
| Waist-to-hip ratio | 0.48 | 0.64 | 0.52 | 0.51 | 0.56 |
| Skinfold thickness (mm) | |||||
| Triceps | 5.5 | 5 | 16 | 20 | 6 |
| Biceps | 4 | 7 | 8 | 11 | 5 |
| Suprailiac | 9 | 18 | 23 | 24 | 11 |
| Subscapular | 17.5 | 34 | 28 | 52 | 28 |
| Thigh | 3.9 | 6 | 15 | 12 | 4 |
| Calf | 3.7 | 3 | 15 | 15 | 2 |
| DXA scan, fat mass (g) | |||||
| Fat % | 21.3 | 31 | 38.6 | 38.9 | 28.9 |
| Total fat | 12,442 | 22,800 | 21,680 | 15,720 | 17,569 |
| Upper-limb fat | 993 | 2677 | 2757 | 2968 | 1991 |
| Lower-limb fat | 2454 | 4735 | 5383 | 6325 | 3151 |
| Trunk fat | 8040 | 14,446 | 12,734 | 15,452 | 11,601 |
| Visceral fat | 872 | 2070 | 871 | 1023 | 1247 |
| Biochemical features | |||||
| Basal glucose (mg/dL) | 103 | 218 | 67 | 94 | 96 |
| Hemoglobin A1c (%) | 6.2 | 7.2 | 5.3 | 5,.2 | 5.7 |
| Plasma insulin (mIU/l) | 5.9 | ND | 20 | 57.8 | 13 |
| Peptide C (ng/mL) | ND | 2.1 | ND | 3,4 | 2,2 |
| Plasma leptin (ng/mL) | 2.4 | 9.7 | ND | 13 | 3,3 |
| Total cholesterol (mg/dL) | 146 | 174 | 264 | 220 | 161 |
| Plasma triglycerides (mg/dL) | 372 | 247 | 214 | 338 | 151 |
| High-density lipoprotein cholesterol (HDLc) (mg/dL) | 26 | 36 | 53 | 39 | 45 |
| Low-density lipoprotein cholesterol (LDLc) (mg/dL) | 39 | 89 | 169 | 113 | 86 |
| Alanine aminotransferase (ALT) (IU/L) | 27 | 56 | 13 | 44 | 26 |
| Aspartate aminotransferase (AST) (IU/L) | 16 | 65 | 16 | 26 | 20 |
| Gamma-glutamyl transpeptidase (GGT) (UI/L) | 9 | 57 | 23 | 23 | 14 |
| Creatinine (mg/dL) | 0.81 | 0.5 | 0.5 | 0.59 | 0.64 |
| Creatine kinase (CK) | ND | 103 | 54 | 81 | 110 |
| Thyroid stimulating hormone (TSH) | 1.43 | 2.02 | 2.78 | 3.27 | 3.58 |
| Blood pressure (BP) | 136/79 | 147/80 | 125/69 | 135/89 | 155/87 |
| Echocardiogram (ECHO) | - | normal | - | normal | mitral regurgitation |
| Medication | Sitagliptin, Fenofibrate, Omega-3 fatty acids, Telmisartan | Metformin, Insulin, Ramipril, Rosuvastatin, Aspirin, Dapaglifozin | - | Metformin | Lormetazepam, Clopidogrel, Aldactone, Atorvastatin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo-Vilar, D.; Fernández-Pombo, A.; Victoria, B.; Mosquera-Orgueira, A.; Cobelo-Gómez, S.; Castro-Pais, A.; Hermida-Ameijeiras, Á.; Loidi, L.; Sánchez-Iglesias, S. Variable Expressivity and Allelic Heterogeneity in Type 2 Familial Partial Lipodystrophy: The p.(Thr528Met) LMNA Variant. J. Clin. Med. 2021, 10, 1497. https://doi.org/10.3390/jcm10071497
Araújo-Vilar D, Fernández-Pombo A, Victoria B, Mosquera-Orgueira A, Cobelo-Gómez S, Castro-Pais A, Hermida-Ameijeiras Á, Loidi L, Sánchez-Iglesias S. Variable Expressivity and Allelic Heterogeneity in Type 2 Familial Partial Lipodystrophy: The p.(Thr528Met) LMNA Variant. Journal of Clinical Medicine. 2021; 10(7):1497. https://doi.org/10.3390/jcm10071497
Chicago/Turabian StyleAraújo-Vilar, David, Antía Fernández-Pombo, Berta Victoria, Adrián Mosquera-Orgueira, Silvia Cobelo-Gómez, Ana Castro-Pais, Álvaro Hermida-Ameijeiras, Lourdes Loidi, and Sofía Sánchez-Iglesias. 2021. "Variable Expressivity and Allelic Heterogeneity in Type 2 Familial Partial Lipodystrophy: The p.(Thr528Met) LMNA Variant" Journal of Clinical Medicine 10, no. 7: 1497. https://doi.org/10.3390/jcm10071497