
Diversity of Pathological Conditions Affecting Pituitary Stalk

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Imaging
2.2. Biochemical Assessment
2.3. Statistics
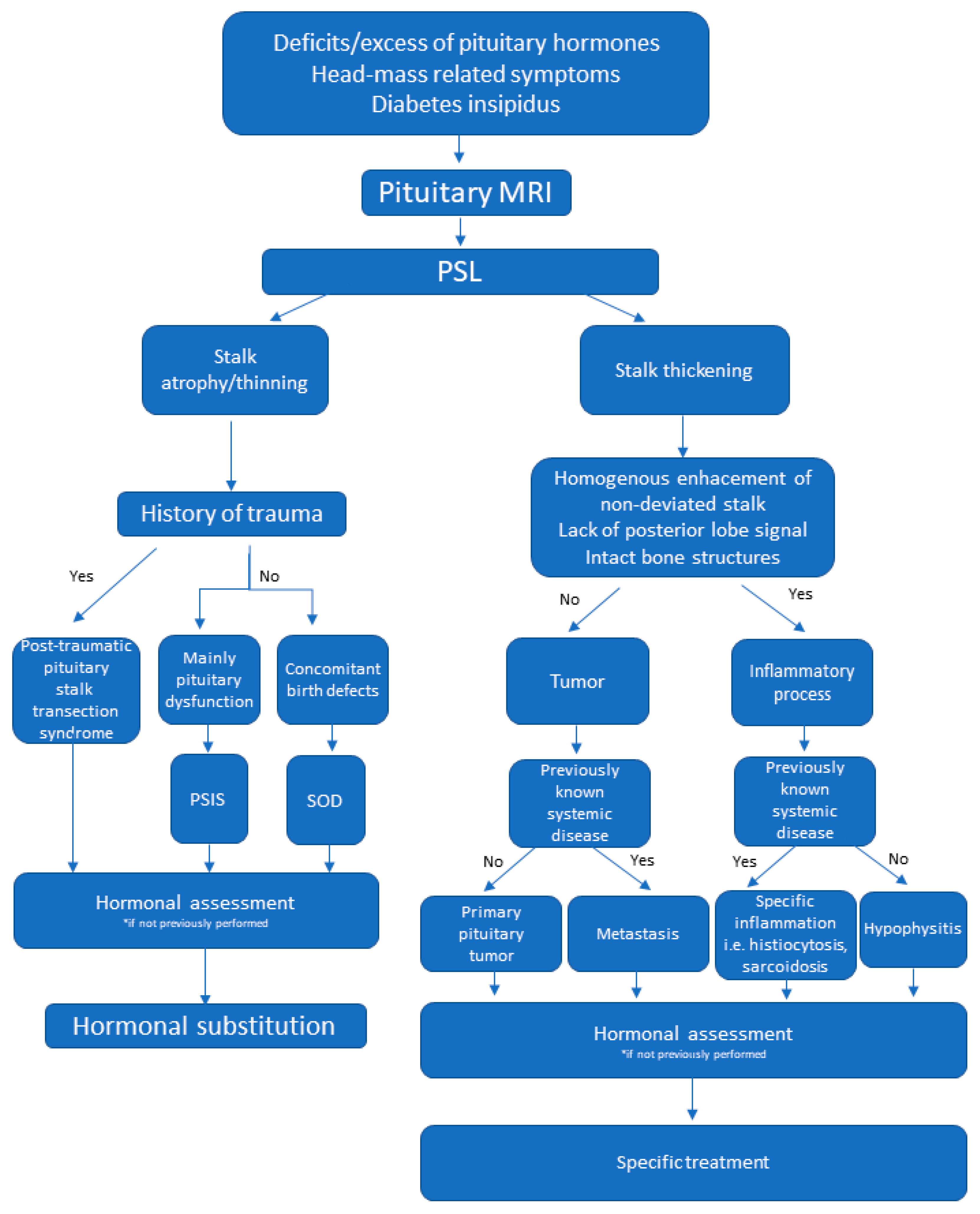
3. Results
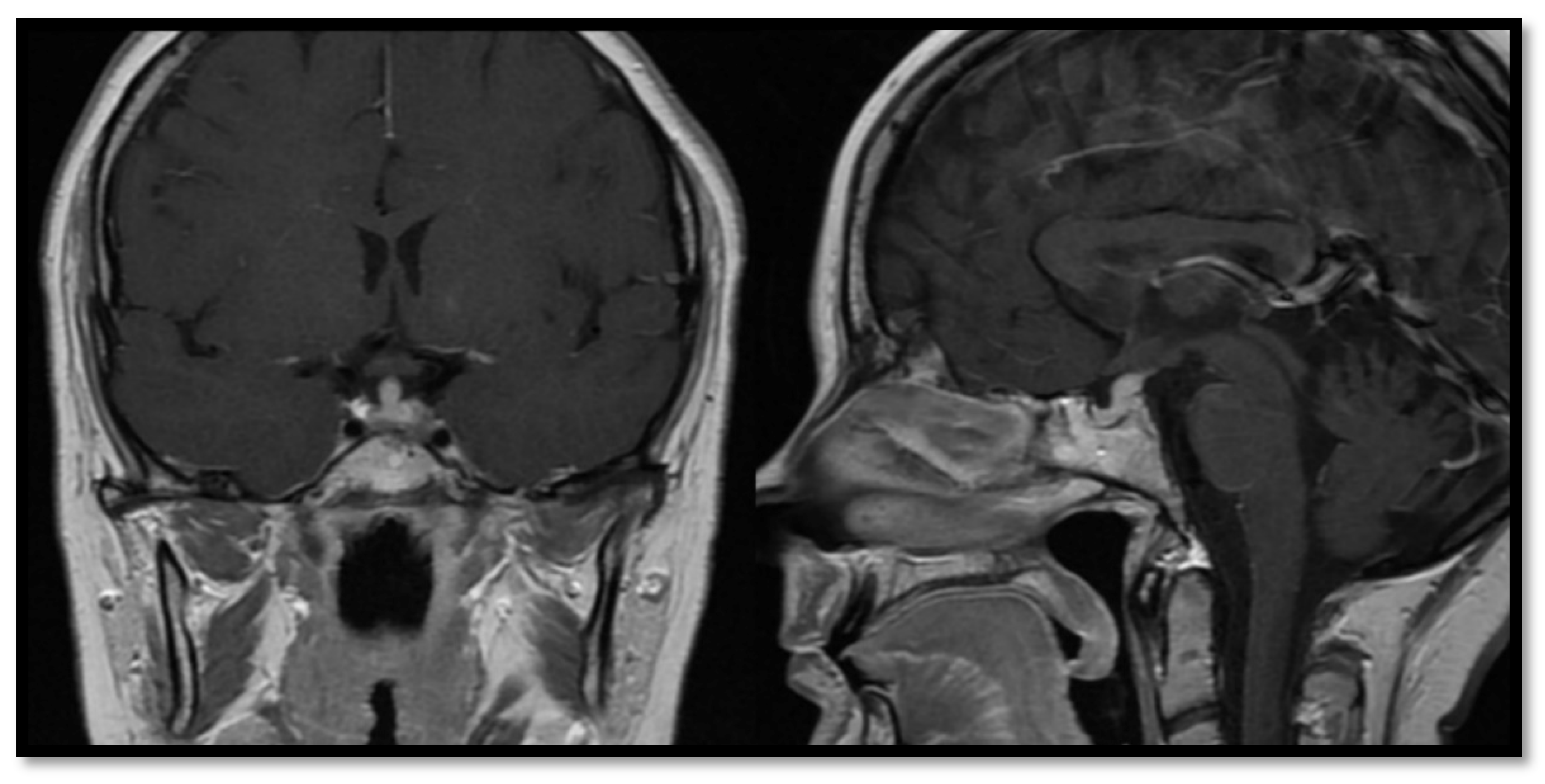
3.1. Congenital Abnormalities
3.2. Inflammatory Processes
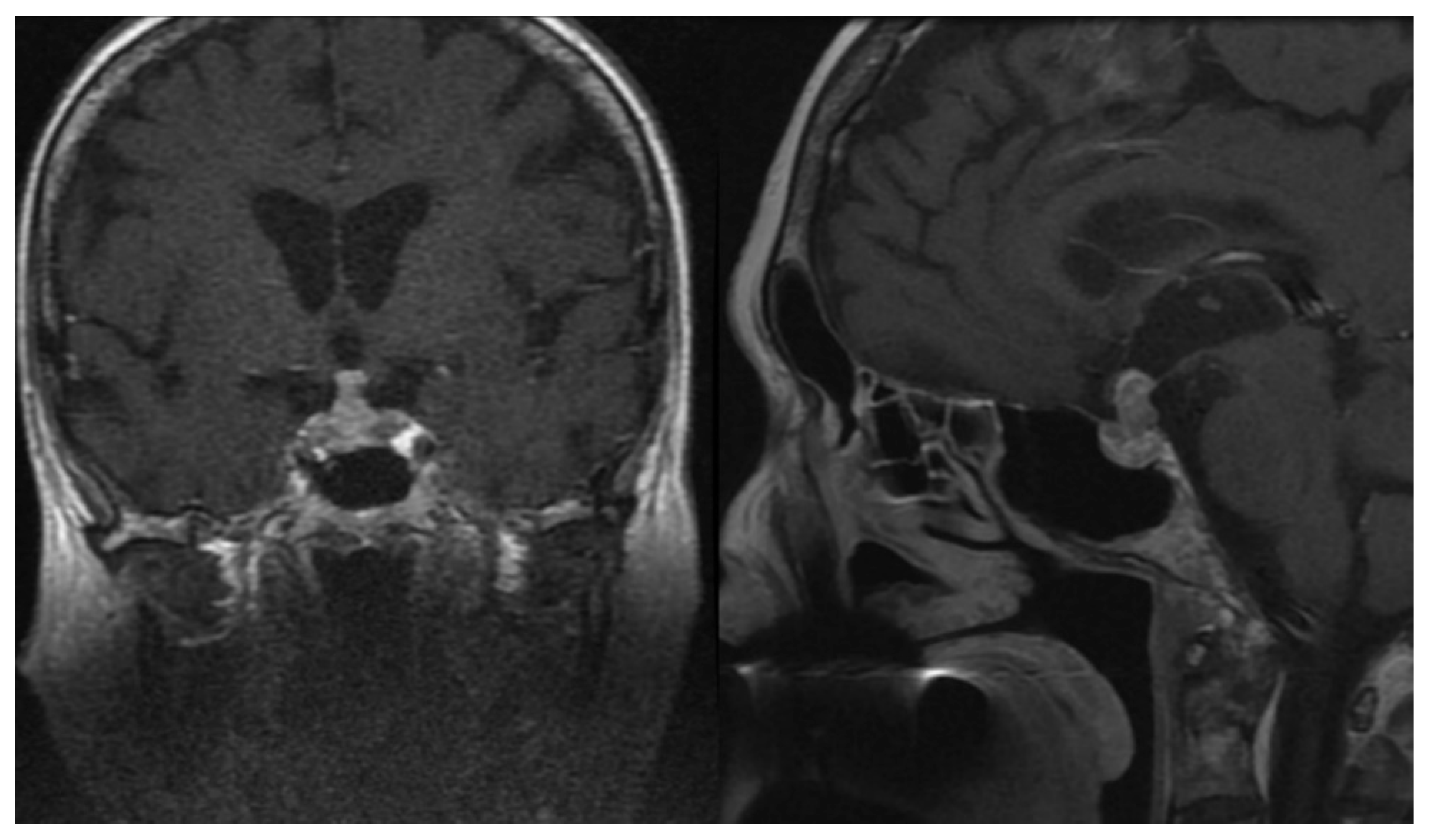
3.3. Neoplastic Change
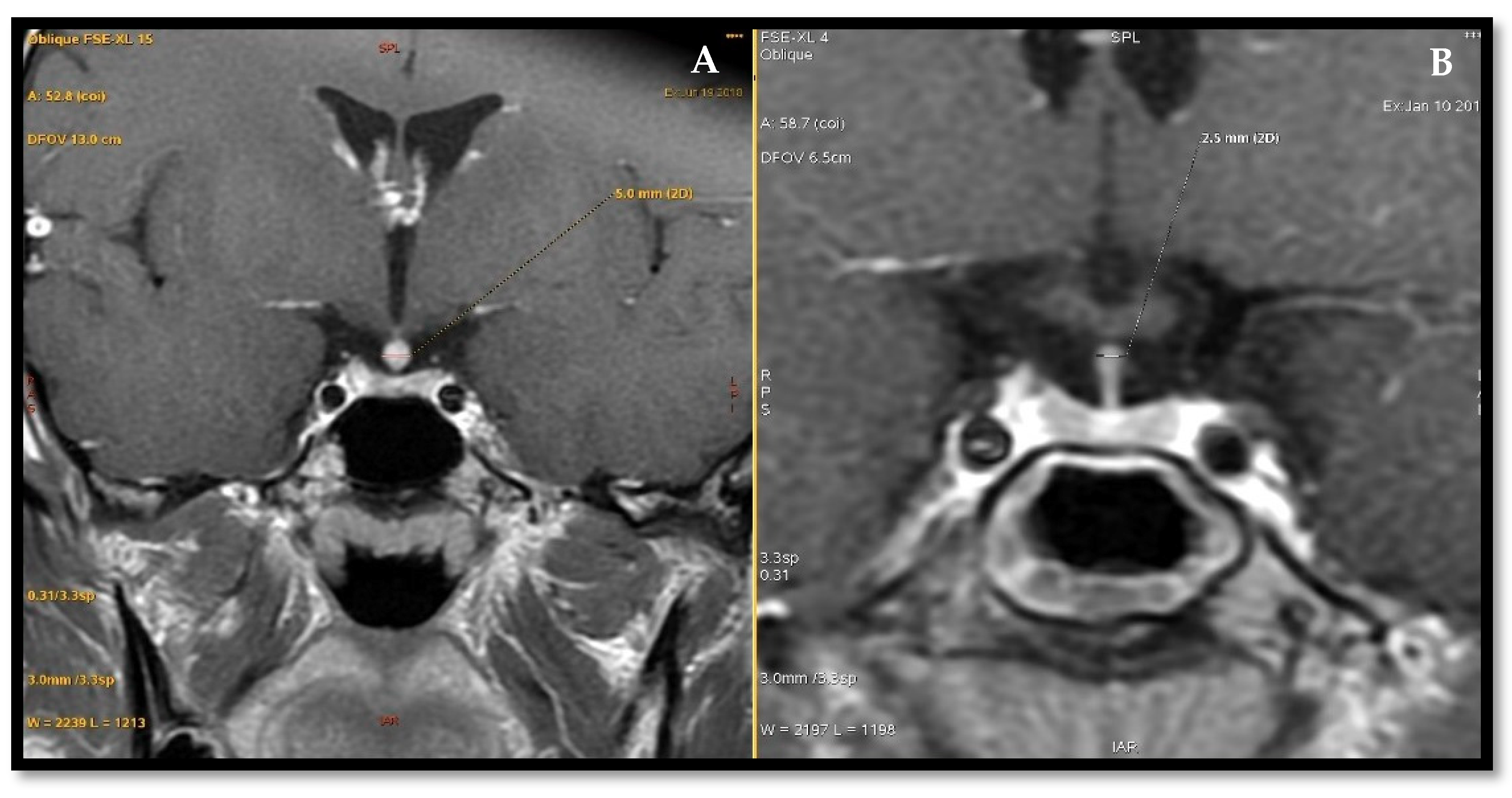
3.4. Imaging
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rupp, D.; Molitch, M. Pituitary stalk lesions. Curr. Opin. Endocrinol. Diabetes Obes. 2008, 15, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Wijetilleka, S.; Khan, M.; Mon, A.; Sharma, D.; Joseph, F.; Sinha, A.; Das, K.; Vora, J. Cranial diabetes insipidus with pituitary stalk lesions. QJM 2016, 109, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doknic, M.; Miljic, D.; Pekic, S.; Stojanovic, M.; Savic, D.; Manojlovic-Gacic, E.; Milenkovic, T.; Zdravkovic, V.; Jesic, M.; Damjanovic, D.; et al. Single center study of 53 consecutive patients with pituitary stalk lesions. Pituitary 2018, 21, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.E.; Salzman, K.L.; Osborn, A.G. Anatomic and pathologic spectrum of pituitary infundibulum lesions. AJR Am. J. Roentgenol. 2007, 188, W223–W232. [Google Scholar] [CrossRef] [PubMed]
- Satogami, N.; Miki, Y.; Koyama, T.; Kataoka, M.; Togashi, K. Normal pituitary stalk: High-resolution MR imaging at 3T. Am. J. Neuroradiol. 2010, 31, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Maintz, D.; Benz-Bohm, G.; Gindele, A.; Schönau, E.; Pfäffle, R.; Lackner, K. Posterior Pituitary Ectopia: Another Hint Toward a Genetic Etiology. Am. J. Neuroradiol. 2000, 21, 1116–1118. [Google Scholar] [PubMed]
- Gutch, M.; Kumar, S.; Razi, S.; Saran, S.; Gupta, K. Pituitary stalk interruption syndrome: Case report of three cases with review of literature. J. Pediatr. Neurosci. 2014, 9, 188. [Google Scholar] [CrossRef]
- Webb, E.A.; Dattani, M.T. Septo-optic dysplasia. Eur. J. Hum. Genet. 2010, 18, 393–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaltsas, G.A.; Powles, T.B.; Evanson, J.; Plowman, P.N.; Drinkwater, J.E.; Jenkins, P.J.; Monson, J.P.; Besser, G.M.; Grossman, A.B. Hypothalamo-Pituitary Abnormalities in Adult Patients with Langerhans Cell Histiocytosis: Clinical, Endocrinological, and Radiological Features and Response to Treatment. J. Clin. Endocrinol. Metab. 2000, 85, 1370–1376. [Google Scholar] [CrossRef]
- Makras, P.; Samara, C.; Antoniou, M.; Zetos, A.; Papadogias, D.; Nikolakopoulou, Z.; Andreakos, E.; Toloumis, G.; Kontogeorgos, G.; Piaditis, G.; et al. Evolving radiological features of hypothalamo-pituitary lesions in adult patients with Langerhans cell histiocytosis (LCH). Neuroradiology 2006, 48, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Zee, C.S.; Go, J.L.; Kim, P.E.; Mitchell, D.; Ahmadi, J. Imaging of the pituitary and parasellar region. Neurosurg. Clin. N. Am. 2003, 14, 55–80. [Google Scholar] [CrossRef]
- Gilis-Januszewska, A.; Kluczyński, Ł.; Rogoziński, D.; Hubalewska-Dydejczyk, A. Radiological and hormonal improvements in a 22-year-old patient with lymphocytic hypophysitis—The watchful waiting approach. Endokrynol. Pol. 2020, 71. [Google Scholar] [CrossRef] [Green Version]
- Turcu, A.F.; Erickson, B.J.; Lin, E.; Guadalix, S.; Schwartz, K.; Scheithauer, B.W.; Atkinson, J.L.D.; Young, W.F. Pituitary stalk lesions: The mayo clinic experience. J. Clin. Endocrinol. Metab. 2013, 98, 1812–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catford, S.; Wang, Y.Y.; Wong, R. Pituitary stalk lesions: Systematic review and clinical guidance. Clin. Endocrinol. 2016, 85, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Shou, X.; Zhang, Z.; Ye, H.; Zhao, Y.; Wang, Y.; Xie, R.; Li, S.; Li, Y. Clinical features of patients with pituitary stalk thickening: A review of 159 cases from one medical center. Chin. Neurosurg. J. 2017, 3, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, X.; Dong, L.; Zhu, B.; Xin, T. MRI features of growth hormone deficiency in children with short stature caused by pituitary lesions. Exp. Ther. Med. 2017, 13, 3474–3478. [Google Scholar] [CrossRef] [Green Version]
- Maghnie, M.; Lindberg, A.; Koltowska-Häggström, M.; Ranke, M.B. Magnetic resonance imaging of CNS in 15,043 children with GH deficiency in KIGS (Pfizer International Growth Database). Eur. J. Endocrinol. 2013, 168, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Deal, C.; Hasselmann, C.; Pfäffle, R.W.; Zimmermann, A.G.; Quigley, C.A.; Child, C.J.; Shavrikova, E.P.; Cutler, G.B.; Blum, W.F. Associations between pituitary imaging abnormalities and clinical and biochemical phenotypes in children with congenital growth hormone deficiency: Data from an international observational study. Horm. Res. Paediatr. 2013, 79, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.-H.; Wang, C.-Z.; Wu, Z.-Q.; Qin, Y.; Han, B.-Y.; Wang, A.-P.; Wang, B.-A.; Dou, J.-T.; Wu, X.-S.; Mu, Y.-M. Multi-genic pattern found in rare type of hypopituitarism: A whole-exome sequencing study of Han Chinese with pituitary stalk interruption syndrome. J. Cell. Mol. Med. 2017, 21, 3626–3632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, J.E.; Muenke, M. Multiple hits during early embryonic development: Digenic diseases and holoprosencephaly. Am. J. Hum. Genet. 2002, 71, 1017–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruszała, A.; Wójcik, M.; Krystynowicz, A.; Starzyk, J.B. Distinguishing between post-trauma pituitary stalk disruption and genetic pituitary stalk interruption syndrome—Case presentation and literature overview. Pediatr. Endocrinol. Diabetes Metab. 2019, 25, 155–162. [Google Scholar] [CrossRef]
- Allen, C.E.; Merad, M.; McClain, K.L. Langerhans-cell histiocytosis. N. Engl. J. Med. 2018, 379, 856–868. [Google Scholar] [CrossRef]
- Leonidas, J.C.; Guelfguat, M.; Valderrama, E. Langerhans’ cell histiocytosis. Lancet 2003, 361, 1293–1295. [Google Scholar] [CrossRef]
- Grois, N.; Pötschger, U.; Prosch, H.; Minkov, M.; Arico, M.; Braier, J.; Henter, J.-I.; Janka-Schaub, G.; Ladisch, S.; Ritter, J.; et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr. Blood Cancer 2006, 46, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Prosch, H.; Grois, N.; Prayer, D.; Waldhauser, F.; Steiner, M.; Minkov, M.; Gadner, H. Central diabetes insipidus as presenting symptom of Langerhans cell histiocytosis. Pediatr. Blood Cancer 2004, 43, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Makras, P.; Alexandraki, K.I.; Chrousos, G.P.; Grossman, A.B.; Kaltsas, G.A. Endocrine manifestations in Langerhans cell histiocytosis. Trends Endocrinol. Metab. 2007, 18, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Tritos, N.A.; Byrne, T.N.; Wu, C.L.; Klibanski, A. A patient with diabetes insipidus, anterior hypopituitarism and pituitary stalk thickening. Nat. Rev. Endocrinol. 2011, 7, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Caturegli, P.; Newschaffer, C.; Olivi, A.; Pomper, M.G.; Burger, P.C.; Rose, N.R. Autoimmune hypophysitis. Endocr. Rev. 2005, 26, 599–614. [Google Scholar] [CrossRef] [Green Version]
- Buxton, N.; Robertson, I.; Powell, M.; Chatterjee, K. Lymphocytic and granulocytic hypophysitis: A single centre experience. Br. J. Neurosurg. 2001, 15, 242–246. [Google Scholar] [CrossRef]
- Prete, A.; Salvatori, R. Hypophysitis—Endotext—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519842/ (accessed on 13 May 2020).
- Honegger, J.; Schlaffer, S.; Menzel, C.; Droste, M.; Werner, S.; Elbelt, U.; Strasburger, C.; Störmann, S.; Küppers, A.; Streetz-van der Werf, C.; et al. Diagnosis of Primary Hypophysitis in Germany. J. Clin. Endocrinol. Metab. 2015, 100, 3841–3849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezzat, S.; Asa, S.L.; Couldwell, W.T.; Barr, C.E.; Dodge, W.E.; Vance, M.L.; McCutcheon, I.E. The prevalence of pituitary adenomas: A systematic review. Cancer 2004, 101, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Osorio, D.S.; Allen, J.C. Management of CNS germinoma. CNS Oncol. 2015, 4, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Karavitaki, N.; Cudlip, S.; Adams, C.B.T.; Wass, J.A.H. Craniopharyngiomas. Endocr. Rev. 2006, 27, 371–397. [Google Scholar] [CrossRef] [PubMed]
- Salge-Arrieta, F.J.; Carrasco-Moro, R.; Rodríguez-Berrocal, V.; Pian, H.; Martínez-San Millán, J.S.; Iglesias, P.; Ley-Urzáiz, L. Clinical features, diagnosis and therapy of pituicytoma: An update. J. Endocrinol. Investig. 2019, 42, 371–384. [Google Scholar] [CrossRef]
- Lieberman, K.A.; Wasenko, J.J.; Schelper, R.; Swarnkar, A.; Chang, J.K.; Rodziewicz, G.S. Tanycytomas: A newly characterized hypothalamic-suprasellar and ventricular tumor. AJNR Am. J. Neuroradiol. 2003, 24, 1999–2004. [Google Scholar] [PubMed]
- Shin, J.H.; Lee, H.K.; Choi, C.G.; Suh, D.C.; Kim, C.J.; Hong, S.K.; Na, D.G. MR Imaging of Central Diabetes Insipidus: A Pictorial Essay. Korean J. Radiol. 2001, 2, 222–230. [Google Scholar] [CrossRef]
- Javanbakht, A.; D’Apuzzo, M.; Badie, B.; Salehian, B. Pituitary metastasis: A rare condition. Endocr. Connect. 2018, 7, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, M.-F.; Brock, M.; Patt, S. Pituitary metastases. MIN Minim. Invasive Neurosurg. 1990, 33, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Nussey, S.; Whitehead, S. The Pituitary Gland—Endocrinology—NCBI Bookshelf. Available online: https://www.ncbi.nlm.nih.gov/books/NBK27/ (accessed on 14 May 2020).
- He, W.; Chen, F.; Dalm, B.; Kirby, P.A.; Greenlee, J.D.W. Metastatic involvement of the pituitary gland: A systematic review with pooled individual patient data analysis. Pituitary 2015, 18, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Bian, L.; Sun, S.; Yang, J.; Chen, X.; Chen, Y.; Ma, Q.; Miao, F.; Wang, W.; Ning, G.; et al. Surgical biopsies in patients with central diabetes insipidus and thickened pituitary stalks. Endocrine 2014, 47, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Jinguji, S.; Nishiyama, K.; Yoshimura, J.; Yoneoka, Y.; Harada, A.; Sano, M.; Fujii, Y. Endoscopic biopsies of lesions associated with a thickened pituitary stalk. Acta Neurochir. 2013, 155, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Mccarthy, B.J.; Shibui, S.; Kayama, T.; Miyaoka, E.; Narita, Y.; Murakami, M.; Matsuda, A.; Matsuda, T.; Sobue, T.; Palis, B.E.; et al. Primary CNS germ cell tumors in Japan and the United States: An analysis of 4 tumor registries. Neuro-Oncology 2012, 14, 1194–1200. [Google Scholar] [CrossRef]
- Hlatky, R.; Suki, D.; Sawaya, R. Carcinoid metastasis to the brain. Cancer 2004, 101, 2605–2613. [Google Scholar] [CrossRef]
- Gur, C.; Lalazar, G.; Salmon, A.; Dubiner, V.; Gross, D.J. Metastatic pancreatic neuroendocrine tumor presenting as a pituitary space occupying lesion: A case report. Pituitary 2008, 11, 293–297. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Congenital (n = 17) | Inflammatory (n = 15) | Neoplastic (n = 20) |
|---|---|---|
| Pituitary stalk interruption syndrome (14) | Lymphocytic hypophysitis (11) | Adenoma (9) |
| Septo-optic dysplasia (2) | Langerhans histiocytosis (4) | Craniopharyngioma (4) |
| Ectopic posterior lobe (1) | Metastatic tumours (3) | |
| Germinoma (1) | ||
| Pituitary carcinoma (1) | ||
| Pituicytoma (1) | ||
| Granular cell tumour (1) |
| Adrenal | Thyroid | Gonadal | Somatotropic | Prolactin | Antidiuretic Hormone | |
|---|---|---|---|---|---|---|
| Deficiency | 19 | 27 | 30 | 24 | 2 | 14 |
| Normal/high | 41 | 33 | 29 | 23 | 39 | 46 |
| High—18 | ||||||
| No data | 0 | 0 | 1 | 13 | 1 | 0 |
| Number | Patient | Sex | Age of Onset | Diagnosis | Symptoms | Cause | Hormonal Function (1—Preserved, 0—Deficiency, H—High Values) | TSH (uIU/mL) | fT4 (pmol/L) | Cortisol 8:00 (ug/dL) | Max. cortisol in 1 ug Synacthen Stimulation (ug/dL) | ACTH (pg/mL) | PRL 8:00 (uIU/mL) | IGF-1 (ng/mL) | LH (mIU/mL) | FSH (mIU/mL) | Testosterone (nmol/L) | Estradiol (pmol/L) | 1st MRI (mm) | 2nd MRI (mm) | Comments | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTH | GH/IGF1 | TSH | LH/FSH | PRL | ADH | 0.27–4.20 | 12.0–22.0 | 2.3–23.3 | 15–65 | Norms in Brackets | Norms in Brackets | F 2.4–12.6—Folicular Phase, >7.7 Menopause; M 1.7–8.6 | F 3.5–12.5—Folicular Phase, >25.8 Menopause; M 1.5–12.4 | 6.68–25.7 or Norms in Brackets | F 46–607—Folicular Phase; 18.4–201 Menopause | |||||||||||
| 1 | AM | F | 14 | Confirmed | growth retardation | PSIS | 0 | 0 | 0 | 0 | 1 H | 1 | 0.02 | 22.31 | 5.77 | x | 18.4 | 3614 (102–496) | 28 (191–478) | <0.1 | <0.1 | x | 590.3 | atrophic | ||
| 2 | MC | F | 3 | growth retardation | 0 | 0 | 0 | 0 | 1 H | 1 | 3.12 | 10.18 | 2.64 | 5.99 | 40.2 | 660 (102–496) | 31 (191–478) | <0.1 | 0.27 | x | 41.04 | atrophic | ||||
| 3 | MP | F | 4 | growth retardation | 0 | 0 | 0 | 0 | 1 | 1 | <0.005 | 21.15 | 1.86 | x | 19 | 480 (102–496) | 33 (191–478) | <0.1 | 0.13 | x | 186.7 | atrophic | ||||
| 4 | KC | M | 3 | cryptorchidism | 1 | 0 | 0 | 0 | 1 H | 1 | 1.14 | 11.82 | 7.48 | 20.79 | 36.3 | 372 (86–324) | 186 (235–408) | 4.17 | 1.42 | 7.36 | x | atrophic | ||||
| 5 | AB | M | 9 | growth retardation | 0 | 0 | 0 | 0 | 1 | 1 | 0.011 | 14.3 | 2.12 | 8.63 | 581 | 128 (102–496) | 96 (160–318) | 0.3 | 0.3 | 1.02 | x | atrophic | High IGF-1: probably laboratory error | |||
| 6 | MO | M | 11 | growth retardation | 1 | 0 | 0 | 0 | 1 H | 1 | 1.71 * | 14.8 * | 9.76 | 23.5 | 61.7 | 472 (86–324) | 122 (235–408) | 1.23 | 1.32 | 16.2 † | x | atrophic | ||||
| 7 | MKn | M | 2 | growth retardation | 1 | 0 | 0 | 1 | 1 H | 1 | 2.57 * | 23.78 * | 12.98 | 21.72 | 17.1 | 442 (86–324) | 137 (235–408) | 4.89 | 2.73 | 25.31 | x | atrophic | ||||
| 8 | PP | M | 11 | growth retardation | 0 | 0 | 0 | 0 | 1 | 1 | 0.01 | 19.84 | <0.05 | x | 12.4 | 97 (86–324) | 26 (235–408) | 0.23 | 0.64 | 14.17 † | x | atrophic | ||||
| 9 | SG | M | 6 | growth retardation | 1 | 0 | 1 | 0 | 1 | 1 | 1.12 | 18.53 | 16.92 | 24.07 | 73.3 | 218 (102–496) | 65 (154–270) | 1.07 | 1.1 | 1.08 | x | atrophic | ||||
| 10 | ŁC | M | 6 | growth retardation | 0 | 0 | 0 | 0 | 1 | 1 | <0.005 | 28.2 | 4.7 | 12.93 | 12 | 309 (102–496) | 219 (154–270) | <0.1 | 0.35 | 16.12 † | x | atrophic | High IGF-1: probably laboratory error, patient after GH therapy | |||
| 11 | KK | M | 9 | growth retardation | 1 | 0 | 0 | 1 | 1 | 1 | 1.9 | 7.72 | 10.14 | x | 34.9 | 80 (30–414) | 150 (154–270) | 4.1 | 5.2 | 3.9 (2.6–10.9) | x | atrophic | ||||
| 12 | SR | M | 19 | growth retardation | 0 | 0 | 0 | 0 | 0 | 1 | <0.005 | 25.6 | 0.2 | x | 6 | 69 (102–496) | x | 0.9 | 1.7 | 9.7 | x | atrophic | Lack of IGF-1 result, but patient after GH therapy | |||
| 13 | SD | M | 1 | hypoglycemia | 0 | 0 | 0 | 0 | 1 | 0 | <0.02 | 12.5 | 0.22 | x | x | 31 (30–414) | 223 (235–408) | x | x | 5.97 | x | atrophic | ||||
| 14 | NS | F | 8 | growth retardation | 0 | 0 | 0 | 0 | 1 | 1 | 2.17 | 10.6 | 3.3 | 10.53 | 12 | 81 (102–496) | 168 (191–478) | <0.1 | 0.37 | x | <18.35 | atrophic | ||||
| 15 | PG | F | 2 | growth retardation | SOD | 0 | 0 | 0 | 0 | 1 | 1 | 0.747 | 11.5 | 1.74 | x | 25.3 | 457 (102–496) | 70 (191–478) | 5.21 | 5.9 | x | 82 | atrophic | |||
| 16 | EB | M | 13 | growth retardation | 1 | 0 | 1 | 1 | 1 | 1 | 1.17 | 18.8 | 14.7 | x | 24.4 | 132 (102–496) | 417 (235–408) $ | 3.04 | 8.17 | 6.85 | x | atrophic | ||||
| 17 | KP | F | 16 | Probable | growth retardation | Ectopic posterior lobe | 1 | 0 | 1 | 0 | 1 | 1 | 2.5 | 14 | 11.39 | x | x | 65 (50–800) | x | 2 | 3 | x | <5.0 | 3 | 3 | Lack of IGF-1 result, patient diagnosed with pituitary dwarfism |
| Number | Patient | Sex | Age of Onset | Diagnosis | Symptoms | Cause | Hormonal Function (1—Preserved, 0—Deficiency, H—High Values) | TSH (uIU/mL) | fT4 (pmol/L) | Cortisol 8:00 (ug/dL) | Max. cortisol in 1 ug Synacthen stimulation (ug/dL) | ACTH (pg/mL) | PRL 8:00 (uIU/mL) | IGF-1 (ng/mL) | LH (mIU/mL) | FSH (mIU/mL) | Testosterone (nmol/L) | Estradiol (pmol/L) | 1st MRI (mm) | 2nd MRI (mm) | 3rd MRI (mm) | 4th MRI (mm) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTH | GH/IGF1 | TSH | LH/FSH | PRL | ADH | 0.27–4.20 | 12.0–22.0 | 2.3–23.3 | 15–65 | Norms in Brackets | Norms in Brackets | F 2.4–12.6—Follicular Phase, >7.7 Menopause; M 1.7–8.6 | F 3.5–12.5—Follicular Phase, >25.8 Menopause; M 1.5–12.4 | 6.68–25.7 | F 46–607—Follicular Phase; 18.4–201 Menopause | ||||||||||||
| 18 | IS | F | 60 | Confirmed | hypopituitarism | Lymphocytic hypophysitis | 0 | 0 | 0 | 0 | 1 | 1 | 0.02 | 11.7 | 1.66 | x | 17 | 140 (102–496) | 73 (122–327) | 1.4 | 4.2 | x | x | 18 × 15 × 12 | 15 | 3 * | |
| 19 | RK | M | 23 | Probable | DI | 1 | 0 | 1 | 1 | 1 | 0 | 3.67 | 16.65 | 15.05 | 30.66 | 39.2 | 384 (102–496) | 225 (235–408) | 5.34 | 2.75 | 9.34 | x | 4.5 | 4.5 | 5 × 5 × 5 | 2.5 × 3 × 3 | |
| 20 | KP | F | 67 | Probable | hypopituitarism | 0 | 1 | 0 | 0 | 1 | 1 | 0.14 | 7.47 | 0.85 | x | <5.0 | 300 (102–496) | 105 (91–320) | x | 6.1 | x | <18.35 | 3.5 | 3.5 | 2.5 | 2.2 | |
| 21 | BI | M | 62 | Probable | DI | 1 | 1 H † | 1 | 1 | 1 H | 0 | 2.19 | 15.47 | 8.81 | 19.09 | 25.8 | 422 (86–324) | 304 (94–245) | 3.84 | 5.47 | 9.44 | x | 4 | 4 | thickened | ||
| 22 | MG | F | 27 | Probable | headache | 1 | 1 | 1 | 1 | 1 H | 1 | 2.71 | 17.88 | 19.09 | x | 22.6 | 598 (102–496) | 287 (191–478) | 16.4 | 6.27 | x | 191 | 4 | 4 | 3 | ||
| 23 | EA | F | 51 | Probable | DI | 1 | 1 | 1 | 1 | 1 | 0 | 0.92 | 12.76 | 11.85 | x | 21.6 | 417 (102–496) | 158 (122–237) | 26.94 | 9.89 | x | <18.35 | 5.5 | 5.5 | 3.5 | 3 | |
| 24 | MK | F | 35 | Probable | hypopituitarism | 1 | x | 0 | 1 * | 1 | 1 | 1.62 | 11.7 | 12.57 | x | 30 | 149 (102–496) | x | 0.52 | 0.99 | x | 467.9 | 3 × 3.5 × 4 | 3.5 | |||
| 25 | TW | M | 71 | Probable | headache | 1 | x | 1 | 1 | 1 | 1 | 3.90 | 18.58 | 24.26 | x | 39.2 | 84 (50–800) | x | 4.98 | 23.69 | 24.87 | x | thickened | thickened | |||
| 26 | KB | M | 10 | Probable | DI | 1 | x | 1 | 1 | 1 | 0 | 2.51 | 15 | 15.3 | x | x | 252 (102–496) | x | x | x | 18.36 | x | 9 × 6.8 × 4.7 | 6.8 | 8 × 3.6 × 3.5 | 2.5–3 | |
| 27 | JK | F | 47 | Probable | hypopituitarism | 0 | 1 | 1 | 1 | 1 | 1 | 1.10 | 16.92 | 0.75 | x | 14.8 | 85 (50–800) | 178 (123–406) | 5.02 | 5.43 | x | 230.5 | 4 | 4 | |||
| 28 | EK | F | 32 | Probable | excessive hormone production | 1 | 1 | 1 | 1 | 1 | 1 | 2.18 | 14 | 17.46 | x | 27.3 | 238 (102–496) | 401 (180–437) | 14.69 | 3.97 | x | 465 | 2.5 | 2.5 | 2.5 | ||
| 29 | SJ | M | 16 | Probable | DI | Langerhans histiocytosis | 1 | 0 | 1 | 1 | 1 | 0 | 1.68 | 15.37 | 16.25 | x | 68 | 226 (102–496) | 221 (235–408) | 3.54 | 2.55 | 26.17 | x | 4.6 × 4 × 4.6 | 4.6 | 3.6 × 2.4 × 3.7 | |
| 30 | Ekr | F | 7 | Probable | DI | 1 | 0 | 1 | 0 | 1 | 0 | 1.50 | 12.57 | 11.96 | 21.75 | 19.1 | 238 (102–496) | 109 (191–478) | <0.1 | 0.46 | x | 102.6 | 3 | 3 | 4 | 4.6 | |
| 31 | KA | M | 4 | Probable | DI | 1 | 1 | 1 | 1 | 1 | 0 | 1.94 | 17.24 | 21.15 | x | 22.7 | 246 (102–496) | 309 (235–408) | 4.56 | 4.16 | 13.44 | x | thickened | ||||
| 32 | ES | F | 38 | Probable | DI | 0 | x | 1 | 0 | 1 | 0 | 1.53 | 12.5 | 6.44 | x | 17.7 | 369 (102–496) | x | 0.127 | 1.15 | x | 46.37 | thickened | ||||
| Number | Patient | Sex | Age of Onset | Diagnosis | Symptoms | Cause | Hormonal Function (1—Preserved, 0—Deficiency, H—High Values) | TSH (uIU/mL) | fT4 (pmol/L) | Cortisol 8:00 (ug/dL) | Max. Cortisol in 1 ug Synacthen Stimulation (ug/dL) | ACTH (pg/mL) | PRL 8:00 (uIU/mL) | IGF-1 (ng/mL) | LH (mIU/mL) | FSH (mIU/mL) | Testosterone (nmol/L) | Estradiol (pmol/L]) | 1st MRI (mm) | 2nd MRI (mm) | 3rd MRI (mm) | 4th MRI (mm) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTH | GH/IGF1 | TSH | LH/FSH | PRL | ADH | 0.27–4.20 | 12.0–22.0 | 2.3–23.3 | 15–65 | Norms in Brackets | Norms in Brackets | F 2.4–12.6—Folicular Phase, >7.7 Menopause; M 1,7–8,6 | F 3.5–12.5—Folicular Phase, >25.8 Menopause; M 1,5–12,4 | 6.68–25.7 or Norms in Brackets | F 46–607—Folicular Phase; 18.4–201 Menopause | ||||||||||||
| 33 | JC | F | 44 | Confirmed | diabetes insipidus | Pituitary carcinoma | 1 | x | 1 | x | 1 | 0 | 0.98 | 14.3 | 36.96 | x | 31 | 724 (50–800) | x | x | x | x | x | 9 | 9 | 10 × 8 × 9 | 12 × 10 × 11 |
| 34 | BP | M | 30 | hypopituitarism | Germinona | 0 | 0 | 0 | 0 | 1 H | 1 | 1.7 | 7.54 | 0.96 | x | 11 | 696 (102–496) | 123 (235–408) | 1.01 | 0.64 | <0.09 | x | 7 | 7 | |||
| 35 | RS | M | 57 | headache | Craniopharygioma | 1 | x | 0 | 0 | 1 | 1 | 0.9 | 5.9 | 7.2 | 26.2 | 17.4 | 159 (50–800) | x | 0.6 | 1.8 | 1.4 (2.6-10.9) | x | 11 | 11 | |||
| 36 | Mka | M | 51 | hypopituitarism | Prolactinoma | 0 | 1 | 0 | 0 | 1 H | 0 | 0.01 | 14.64 | 0.27 | x | 9.6 | 83648 (86–324) | 212 (144–286) | <0.1 | 0.62 | 0.09 | x | 8 | 7 | 11 | 18 × 14 | |
| 37 | KM | F | 71 | headache | Corticotropinoma | 1 | 0 | 0 | 0 | 1 | 1 | 1.21 | 12.1 | 25.38 | x | 108.7 | 335 (102–496) | 42 (91–320) | 0.3 | 0.3 | x | 135 | 7 × 6 × 5 | 6 | |||
| 38 | AC | F | 21 | excessive horomne production | Corticotropinoma | 1 | 1 | 0 | 0 | 1 | 0 | 0.1 | 15.27 | 7.63 | 18.12 | 38 | 119 (102–496) | 204 (191–478) | <0.1 | <0.1 | x | 587.3 | thickened | ||||
| 39 | MN | F | 25 | excessive horomne production | Acromegaly | 1 | x | 0 | 0 | 1 | 1 | 0.07 | 18 | 19.2 | x | x | 229 (102–496) | x | 3.6 (4–12) | 3.9 | 0.7 | x | 8 | 8 | |||
| 40 | Abo | F | 57 | excessive horomne production | Acromegaly | 1 | 1 H | 1 | 1 | 1 | 1 | 0.76 | 12.8 | 14.86 | x | 15.7 | 324 (102–496) | 538 (122–237) | 20.5 | 66 | x | x | thickened | ||||
| 41 | KZ | F | 28 | headache | Adenoma | 1 | 1 | 1 | 1 | 1 | 1 | 1.48 | 14.18 | 15.56 | x | 57 | 401 (102–496) | 412 (191–478) | 2.94 | 6.46 | x | 123.5 | 21 × 14 × 19 * | ||||
| 42 | Mgu | F | 57 | Probable | hypopituitarism | Prolactinoma | 0 | x | 1 | 0 | 1 H | 1 | 0.19 | 13.4 | 5.7 | x | 25 | 608 (40–470) | x | x | 0.59 | x | x | thickened | |||
| 43 | JKo | F | 63 | confusion | Craniopharyngioma | 1 | 1 | 0 | 0 | 0 | 1 | 0.37 | 9.98 | 8.38 | 20.7 | 18 | 7 † (102–496) | 214 (91–320) | 0.3 | 0.46 | x | 38.57 | 28 × 25 × 18 * | ||||
| 44 | KD | F | 22 | hypopituitarism | Craniopharyngioma | 1 | 0 | 1 | 1 | 1 H | 1 | 1.09 | 15.54 | 23.1 | x | 22.8 | 922 (102–496) | 142 (191–478) | 9.94 | 5.73 | x | 119 | 3 × 5 | 3 | 3.5 | 3.8 | |
| 45 | MS | M | 22 | seizure | Craniopharyngioma | 1 | 1 | 1 | 1 H $ | 1 | 1 | 3.11 | 17.86 | 11.4 | x | 25.8 | 237 (102–496) | 341 (235–408) | 19.44 | 27.91 | 7.09 (8.64–29) | x | 2.3 | 2.3 | 2.3 | ||
| 46 | DZ | F | 61 | incidental | Adenoma | 1 | 1 | 1 | 1 | 1 H | 1 | 2.9 | 14.79 | 19.88 | x | 134 | 690 (102–496) | 116 (91–320) | 21.73 | 35.33 | x | x | 8 × 5 × 3.6 | 5 | 7.5 × 4.6 × 6 | 6 × 4.5 × 6 | |
| 47 | DP | F | 60 | incidental | Pituicytoma | 1 | x | 1 | 1 | 1 | 1 | 2.37 | 13.4 | 12.84 | 24.19 | 18.1 | 286 (102–496) | x | 11.5 (>10) | 53 | x | x | 11 × 8 × 9 | 8 | 11 × 9 × 11 | ||
| 48 | PM | M | 28 | headache | Adenoma | 1 | x | 1 | 1 | 1 | 1 | 12.8 | 13.3 | 11.4 | x | 60 | 117 (102–496) | x | 3 | 4.5 | 3 (2.6–10.9) | x | 7 × 6 × 8 | 8 | 7 × 6 × 8 | ||
| 49 | GP | F | 68 | diabetes insipidus | NET metastasis | 1 | x | 0 | 0 | 1 H | 0 | 0.01 | 12 | 18.4 | x | 15.1 | 887 (102–496) | x | 3.3 | 4 | 3 | x | 6 | 6 | |||
| 50 | MKr | F | 74 | hypopituitarism | NET metastasis | 0 | 1 | 0 | 0 | 1 H | 1 | 0.04 | 8.29 | 0.3 | x | 16.3 | 823 (102–496) | 125 (91–320) | <0.1 | 0.25 | x | 169.4 | 7.5 × 12 × 6 | 7.5 | |||
| 51 | JT | F | 60 | hypopituitarism | Acromegaly +lung cancer metastasis | 0 | 1 H | 1 | 0 | 1 H | 1 | 0.19 | 14.1 | 1.27 | 12.79 | 17.3 | 928 (102–496) | 452 (122–327) | <0.1 | 0.28 | x | x | 4 | 4 | |||
| 52 | MH | M | 35 | diabetes insipidus | Granullar cell tumor | 1 | x | 1 | 1 | 1 | 0 | 1.57 | 12.56 | 20.08 | x | 27.8 | 228 (102–496) | x | 9.25 | 7.36 | 9.44 | x | 3 | 3 | 4 | 4.1 | |
| Number | Patient | Sex | Age of Onset | Aetiology and Diagnosis | Symptoms | Hormonal Function (1—Preserved, 0—Deficiency, H—High Values) | TSH (uIU/mL) | fT4 (pmol/L) | Cortisol 8:00 (ug/dL) | Max. cortisol in 1 ug Synacthen Stimulation (ug/dL) | ACTH (pg/mL) | PRL 8:00 (uIU/mL) | IGF-1 (ng/mL) | LH (mIU/mL) | FSH (mIU/mL) | Testosterone (nmol/L) | Estradiol (pmol/L) | 1st MRI (mm) | 2nd MRI (mm) | 3rd MRI (mm) | 4th MRI (mm) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTH | GH/IGF1 | TSH | LH/FSH | PRL | ADH | 0.27–4.20 | 12.0–22.0 | 2.3–23.3 | 15–65 | Norms in Brackets | Norms in Brackets | F 2.4–12.6—Folicular Phase, >7.7 Menopause; M 1.7–8.6 | F 3.5–12.5—Folicular Phase, >25.8 Menopause; M 1.5–12.4 | 6.68–25.7 or Norms in Brackets | F 46–607—Folicular Phase; 18.4–201 Meno Pause | |||||||||||
| 53 | MB | F | 56 | Undetermined | headache | 1 | 1 | 1 | 1 | 1 | 1 | 1.3 | 18.05 | 36 | x | 82 | 167 (102–496) | 156 (14–286) | 24.49 | 42.12 | x | <18.35 | 15 × 11 × 10 * | 11 | ||
| 54 | EW | M | 58 | headache | 1 | x | 1 | 1 | 1 H | 1 | 0.9 | 16 | 19.3 | x | x | 353 (35–330) | x | 7.9 | 12.5 | x | x | 5 | 5 | |||
| 55 | Mgo | F | 43 | headache | 1 | x | 1 | 1 | x | 1 | x | 14.86 | 17.54 | x | 27 | x | x | 6.92 | 4.48 | x | x | 12 × 11 × 7 | 11 | 12 × 11 × 7 | 11 × 10 × 7 | |
| 56 | Dpa | F | 77 | headache | 1 | 1 | 1 | 1 | 1 | 1 | 1.3 | 16.33 | 24 | x | 112 | 196 (102–496) | 96 (91–320) | 39.02 | 82.7 | x | x | 11 × 7 × 9 | 9 | |||
| 57 | KK | M | 57 | headache | 1 | 1 | 1 | 1 | 1 | 1 | 4.9 | 12.59 | 18 | x | 21.7 | 325 (102–496) | 134 (94–245) | 6.37 | 5.18 | 6.3 (2.6–10.9) | x | 7 × 5 × 7 | 7 | 7 × 5 × 7 | ||
| 58 | Mse | M | 25 | hypopituitarism | 1 | 1 | 1 | 0 | 1 | 1 | 1.9 | 13.57 | 41.7 | x | 29.2 | 449 (102–496) | 339 (235–408) | 0.92 | 0.38 | 0.77 | x | thickened | ||||
| 59 | EM | F | 28 | excessive hormone production | 1 | 1 | 1 | 1 | 1 H | 1 | 1.49 | 14.04 | 27.8 | x | 17.7 | 670 (102–496) | 270 (191–478) | 6.42 | 6.01 | x | 341.8 | 3 | 3 | |||
| 60 | ZS | F | 64 | hypopituitarism | 1 | 1 | 0 | 1 | 1 H | 1 | 1.95 | 8.11 | 19.04 | x | 35 | 827 (102–496) | 192 (91–320) | 15.55 | 31.78 | x | <18.35 | 8 | 8 | 9 × 10 × 8 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kluczyński, Ł.; Gilis-Januszewska, A.; Godlewska, M.; Wójcik, M.; Zygmunt-Górska, A.; Starzyk, J.; Hubalewska-Dydejczyk, A. Diversity of Pathological Conditions Affecting Pituitary Stalk. J. Clin. Med. 2021, 10, 1692. https://doi.org/10.3390/jcm10081692
Kluczyński Ł, Gilis-Januszewska A, Godlewska M, Wójcik M, Zygmunt-Górska A, Starzyk J, Hubalewska-Dydejczyk A. Diversity of Pathological Conditions Affecting Pituitary Stalk. Journal of Clinical Medicine. 2021; 10(8):1692. https://doi.org/10.3390/jcm10081692
Chicago/Turabian StyleKluczyński, Łukasz, Aleksandra Gilis-Januszewska, Magdalena Godlewska, Małgorzata Wójcik, Agata Zygmunt-Górska, Jerzy Starzyk, and Alicja Hubalewska-Dydejczyk. 2021. "Diversity of Pathological Conditions Affecting Pituitary Stalk" Journal of Clinical Medicine 10, no. 8: 1692. https://doi.org/10.3390/jcm10081692
APA StyleKluczyński, Ł., Gilis-Januszewska, A., Godlewska, M., Wójcik, M., Zygmunt-Górska, A., Starzyk, J., & Hubalewska-Dydejczyk, A. (2021). Diversity of Pathological Conditions Affecting Pituitary Stalk. Journal of Clinical Medicine, 10(8), 1692. https://doi.org/10.3390/jcm10081692