
Resistance to Immune Checkpoint Inhibitors Secondary to Myeloid-Derived Suppressor Cells: A New Therapeutic Targeting of Haematological Malignancies


 , , , and
, , , and
Abstract
:1. Introduction
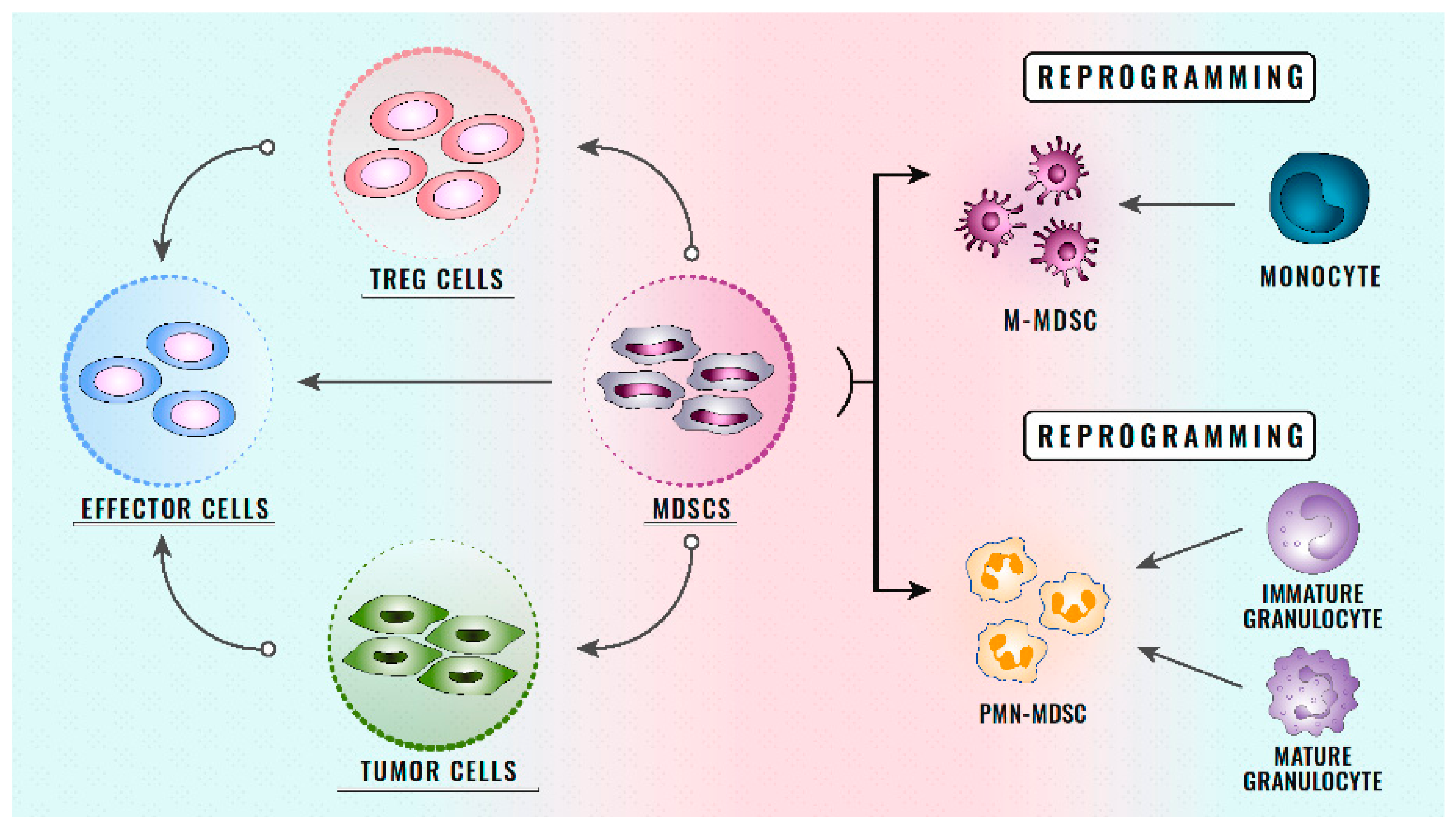
2. Definition and Role of MDSCs
3. Classification of MDSCs
4. MDSCs and Haematological Malignancies
4.1. Lymphoma
4.1.1. MDSCs in Hodgkin’s Lymphoma
4.1.2. MDSCs in Non-Hodgkin’s Lymphoma
4.2. Multiple Myeloma
4.3. Leukaemia
4.3.1. Acute Leukaemias
4.3.2. Myeloproliferative Neoplasms
4.3.3. Chronic Lymphocytic Leukaemia (CLL)
4.4. Myelodisplastic Syndromes
5. Immunotherapy for Haematological Malignancies
5.1. Hodgkin’s Lymphoma
5.2. Non-Hodgkin’s Lymphoma
5.3. Multiple Myeloma
5.4. Leukaemia
6. Resistance to ICIs Secondary to MDSCs
- Direct action on T cells: As the main objective of ICIs, the function of T cells is essential for the response of patients receiving treatment with ICIs, and is affected by MDSCs directly through cell–cell contact, correlating with a poor clinical outcome in patients treated with ICIs [133,134,135,136]. Some studies have shown how, in TME, the MDSCs present a high expression of PD-L1, which reduces the T cell populations in TME. After treatment with immunotherapy, the MDSCs have been observed to begin a high expression of different receptors that generate a negative regulation on the antitumour mechanisms of T cells [137,138] (Figure 1). In the future, more studies are needed to assess how the function of T cells is affected by ICIs secondary to the change in the phenotype of MDSCs.
- Angiogenesis secondary to MDSCs: The myeloid origin of MDSCs carries an ability to facilitate angiogenesis in TME in both solid and haematological tumours [139]. These processes have been observed to be fundamental in angiogenesis through the administration of BV8 in haematological neoplasms [140]. This specific antibody significantly reduced new blood vessels in TME, leading to a key antitumour response. Therefore, antiangiogenic drugs associated with anti-MDSC therapies could be a key point in the treatment of tumours with a high expression of MDSCs in the future [141,142]. Likewise, in the future, the combination of therapies with ICIs plus antiangiogenics and drugs that alter the number or function of MDSCs could be a new way to treat haematological neoplasms. It will be important to assess the toxicity associated with these treatments, since currently antiangiogenic drugs show partial specificity.
- Interaction of MDSCs with TME: Different TME cells can generate greater secondary immunosuppression on contact with MDSCs [143,144,145]. Different pathways, such as Wnt/β-catenin, can interact with MDSC receptors, generating greater tumour leakage. These interactions not only affect the immune system, but can also help angiogenesis, metalloprotease expression, or the epithelium–mesenchyme transition. There are currently various drugs in clinical trials that are directed against the microenvironment in different neoplasms. These drugs will be able to modify the phenotype of the MDSCs and their associated immunosuppression. The combination of these therapies with immunotherapy may make it possible to enhance the activating effect of the immune system through the direct activation of T cells by ICIs, as well as the decrease and alteration of the function of MDSCs.
7. Targeting MDSCs to Overcome Resistance to ICIs
7.1. ICIs and Reduction of the Number of MDSCs
7.2. ICIs and Functional Alteration of MDSCs
8. Future Paths in MDSCs and ICIs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ALL | Acute lymphoblastic leukaemia |
| AML | Acute myeloid leukaemia |
| APL | Acute promyelocytic leukaemia |
| ATRA | All-trans retinoic acid |
| B-ALL | B-acute lymphoblastic leukaemia |
| BTK | Bruton’s tyrosine kinase |
| BV | Brentuximab vedotin |
| cHL | Classical Hodgkin lymphoma |
| CLL | Chronic lymphocytic leukaemia |
| CML | Chronic myeloid leukaemia |
| CR | Complete response |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
| DLBCL | Diffuse large B cell lymphoma |
| DNMTi | DNA methyltransferase inhibitors |
| EBV | Epstein–Barr virus |
| EMA | European Medicines Agency |
| ENKL | Extranodal NK/T cell lymphoma |
| FDA | US Food and Drug Administration |
| FGF | Fibroblast growth factors |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| HL | Hodgkin lymphoma |
| HRS | Hodgkin Reed Sternberg |
| ICI | Immune checkpoint inhibitor |
| IDO | Indoleamine-pyrrole 2,3-dioxygenase |
| IMC | Immature myeloid cell |
| IMiDs | Immunomodulatory imide drugs |
| IPI | International Prognostic Index |
| ITIM | Immunoreceptor tyrosine-based activation motif |
| MDSC | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| M-MDSC | Monocytic myeloid-derived suppressor cells |
| MYC | Myelocytomatosis proto-oncogene |
| NHL | Non-Hodgkin lymphoma |
| NK cells | Natural killer cells |
| NO | Nitrate reductases |
| ORR | Overall response rate |
| PBMC | Peripheral blood mononuclear cells |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PD-L2 | Programmed death-ligand 2 |
| PFS | Progression free survival |
| PIWI | P-element Induced Wimpy testis |
| PMBL | Primary mediastinal B cell lymphoma |
| PMN-MDSC | Polymorphonuclear myeloid-derived suppressor cells |
| PR | Partial response |
| ROS | Reactive oxygen species |
| R/R | Relapse/Refractory |
| T-ALL | T acute lymphoblastic leukaemia |
| TCR | T cell receptor |
| TFG-β | Transforming growth factor beta |
| TIGIT | T cell immunoglobulin and ITIM domain |
| TME | Tumour microenvironment |
| VEGF | Vascular endothelial growth factor |
| VISTA | V-domain immunoglobulin suppressor of T cell activation |
| WNT | Wingless way |
References
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-Derived Suppressor Cells: Not Only in Tumor Immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, E.; Asadzadeh, Z.; Safaei, S.; Hatefi, A.; Derakhshani, A.; Giovannelli, F.; Brunetti, O.; Silvestris, N.; Baradaran, A. MicroRNAs and IncRNAs—A new layer of myeloid-derived suppressor cells regulation. Front. Immunol. 2020, 11, 572323. [Google Scholar] [CrossRef] [PubMed]
- Pastaki Khoshbin, A.; Eskian, M.; Keshavarz-Fathi, M.; Rezaei, N. Roles of Myeloid-Derived Suppressor Cells in Cancer Metastasis: Immunosuppression and Beyond. Arch. Immunol. Ther. Exp. 2019, 67, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, C.; Wang, J.; Xue, L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int. J. Cancer. 2019, 144, 933–946. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; van Valckenborgh, E.E.; Lahmar, Q.; Geeraerts, X.; de Bruyne, E.; Menu, E.; Van Riet, I.; Vanderkerken, K.; Van Ginderachter, J.A. Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front. Oncol. 2014, 4, 349. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, K.; Huang, X.J. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar]
- Qu, P.; Wang, L.Z.; Lin, P.C. Expansion and functions of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Lett. 2016, 380, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar] [PubMed]
- Park, S.M.; Youn, J.I. Role of myeloid-derived suppressor cells in immune checkpoint inhibitor therapy in cancer. Arch. Pharm. Res. 2019, 42, 560–566. [Google Scholar] [CrossRef]
- Toor, S.M.; Elkord, E. Therapeutic prospects of targeting myeloid-derived suppressor cells and immune checkpoints in cancer. Immunol. Cell Biol. 2018, 96, 888–897. [Google Scholar] [CrossRef]
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Sui, H.; Zhao, S.; Gao, X.; Su, Y.; Qu, P. Immunotherapy Targeting Myeloid-Derived Suppressor Cells (MDSCs) in Tumor Microenvironment. Front. Immunol. 2021, 11, 585214. [Google Scholar] [CrossRef]
- Kramer, E.D.; Abrams, S.I. Granulocytic Myeloid-Derived Suppressor Cells as Negative Regulators of Anticancer Immunity. Front. Immunol. 2020, 11, 1963. [Google Scholar] [CrossRef]
- Safarzadeh, E.; Hashemzadeh, S.; Duijf, P.H.G.; Mansoori, B.; Khaze, V.; Mohammadi, A.; Kazemi, T.; Yousefi, M.; Asadi, M.; Mohammadi, H.; et al. Circulating myeloid-derived suppressor cells: An independent prognostic factor in patients with breast cancer. J. Cell Physiol. 2019, 234, 3515–3525. [Google Scholar] [CrossRef]
- Pilatova, K.; Budinská, E.; Bensciková, B.; Nenutil, R.; Šefr, R.; Fedorová, L.; Hanáková, B.; Brychtová, V.; Zdrazilová Dubská, L. Circulating myeloid suppressor cells and their role in tumour immunology. Klinicka Onkologie 2017, 30 (Suppl. S1), 166–169. [Google Scholar]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-derived exosomes, myeloid-derived suppressor cells, and tumor microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Huang, X.; Yang, Y. Myeloid-Derived Suppressor Cells Regulate Natural Killer Cell Response to Adenovirus-Mediated Gene Transfer. J. Virol. 2012, 86, 13689–13696. [Google Scholar] [CrossRef] [Green Version]
- Monu, N.R.; Frey, A.B. Myeloid-derived suppressor cells and anti-tumor T cells: A complex relationship. Immunol. Investig. 2012, 41, 595–613. [Google Scholar] [CrossRef] [Green Version]
- Vetsika, E.K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef]
- Law, A.M.; Lim, E.; Ormandy, C.J.; Gallego-Ortega, D. The innate and adaptive infiltrating immune systems as targets for breast cancer immunotherapy. Endocr. Relat. Cancer 2017, 24, R123–R144. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5489–5505. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Hoechst, B.; Duffy, A.; Gamrekelashvili, J.; Fioravanti, S.; Manns, M.P.; Greten, T.F.; Korangy, F. S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology 2012, 136, 176–183. [Google Scholar] [CrossRef]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Koinis, F.; Gioulbasani, M.; Aggouraki, D.; Koutoulaki, A.; Skalidaki, E.; Mavroudis, D.; Georgoulias, V.; Kotsakis, A. A circulating subpopulation of monocytic myeloid-derived suppressor cells as an independent prognostic/predictive factor in untreated non-small lung cancer patients. J. Immunol. Res. 2014, 2014, 659294. [Google Scholar] [CrossRef]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Trikha, P.; Carson, W.E. Signaling pathways involved in MDSC regulation. Biochim. Biophys. Acta 2014, 1846, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Parrinello, N.L.; Vetro, C.; Forte, S.; Chiarenza, A.; Figuera, A.; Motta, G.; Palumbo, G.A.; Ippolito, M.; Consoli, U.; et al. Circulating myeloid-derived suppressor cells correlate with clinical outcome in Hodgkin lymphoma patients treated up-front with a risk-adapted strategy. Br. J. Haematol. 2015, 168, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-J.; Shively, J.E. The cell-cell adhesion molecule carcinoembryonic antigen-related cellular adhesion molecule 1 inhibits IL-2 production and proliferation in human T cells by association with Src homology protein-1 and down-regulates IL-2 receptor. J. Immunol. 2004, 172, 3544–3552. [Google Scholar] [CrossRef] [Green Version]
- Marini, O.; Spina, C.; Mimiola, E.; Cassaro, A.; Malerba, G.; Todeschini, G.; Perbellini, O.; Scupoli, M.; Carli, G.; Facchinelli, D.; et al. Identification of granulocytic myeloid-derived suppressor cells (G-MDSCs) in the peripheral blood of Hodgkin and non-Hodgkin lymphoma patients. Oncotarget 2016, 7, 27676–27688. [Google Scholar] [CrossRef] [Green Version]
- Amini, R.M.; Enblad, G.; Hollander, P.; Eriksson, E.; Ayoola Gustafsson, K.; Loskog, A.; Thörn, I. Altered profile of immune regulatory cells in the peripheral blood of lymphoma patients. BMC Cancer 2019, 19, 316. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Gustafson, M.P.; Bulur, P.A.; Gastrineau, D.A.; Witzig, T.E.; Dietz, A.B. Immunosuppressive CD14+HLA-DR(low)/- monocytes in B-cell non-Hodgkin lymphoma. Blood 2011, 117, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, K.A.; Badawy, H.M.; Radwan, W.M.; Shehata, M.A.; Bassuoni, M.A. CD14(+) HLA-DR low/(−) monocytes as indicator of disease aggressiveness in B-cell non-Hodgkin lymphoma. Int. J. Lab. Hematol. 2014, 36, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, T.; Fell, R.; Polliack, A.; Attias, D. Absolute monocytosis at diagnosis correlates with survival in diffuse large B-cell lymphoma-possible link with monocytic myeloid-derived suppressor cells. Hematol. Oncol. 2013, 31, 65–71. [Google Scholar] [CrossRef]
- Xiu, B.; Lin, Y.; Grote, D.M.; Ziesmer, S.C.; Gustafson, M.P.; Maas, M.L.; Zhang, Z.; Dietz, A.B.; Porrata, L.F.; Novak, A.J.; et al. IL-10 induces the development of immunosuppressive CD14(+)HLA-DR (low/−) monocytes in B-cell non- Hodgkin lymphoma. Blood Cancer J. 2015, 5, e328. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, Z.-L.; Ye, S.-B.; Ouyang, L.-Y.; Chen, Y.-S.; He, J.; Huang, H.-Q.; Zeng, Y.-X.; Zhang, X.-S.; Li, J. Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: A novel prognostic indicator. Cancer Immunol. Immunother. 2015, 64, 1587–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Wu, X.; Zhang, X.; Chai, Y.; Guo, Q.; Li, L.; Yue, L.; Bai, J.; Wang, Z.; Zhang, L. Prognostic significance of peripheral monocytic myeloid-derived suppressor cells and monocytes in patients newly diagnosed with diffuse large b-cell lymphoma. Int. J. Clin. Exp. Med. 2015, 8, 15173–15181. [Google Scholar] [PubMed]
- Bachanova, V.; Sarhan, D.; DeFor, T.E.; Cooley, S.; Panoskaltsis-Mortari, A.; Blazar, B.R.; Curtsinger, J.M.; Burns, L.; Weisdorf, D.J.; Miller, J.S. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol. Immunother. 2018, 67, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, D.; Cichocki, F.; Zhang, B.; Yingst, A.; Spellman, S.R.; Cooley, S.; Verneris, M.R.; Blazar, B.R.; Miller, J.S. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5696–5706. [Google Scholar] [CrossRef] [Green Version]
- Brimnes, M.K.; Vangsted, A.J.; Knudsen, L.M.; Gimsing, P.; Gang, A.O.; Johnsen, H.E.; Snave, I.M. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR(-)/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand. J. Immunol. 2010, 72, 540–547. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, L.; Wang, H.; Xiong, S.; Li, Y.; Tao, Q.; Xiao, W.; Qin, H.; Wang, Y.; Zhai, Z. Tumor-induced CD14+HLA-DR (-/low) myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol. Immunother. 2015, 64, 389–399. [Google Scholar] [CrossRef]
- Ramachandran, I.R.; Martner, A.; Pisklakova, A.; Condamine, T.; Chase, T.; Vogl, T.; Roth, J.; Gabrilovich, D.; Nefedova, Y. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J. Immunol. 2013, 190, 3815–3823. [Google Scholar] [CrossRef] [Green Version]
- Favaloro, J.; Liyadipitiya, T.; Brown, R.; Yang, S.; Suen, H.; Woodland, N.; Nassif, N.; Hart, D.; Fromm, P.; Weatherburn, C.; et al. Myeloid derived suppressor cells are numerically, functionally and phenotypically different in patients with multiple myeloma. Leuk. Lymphoma 2014, 55, 2893–2900. [Google Scholar] [CrossRef]
- Ai, L.; Mu, S.; Sun, C.; Fan, F.; Yan, H.; Qin, Y.; Cui, G.; Wang, Y.; Guo, T.; Mei, H.; et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 2019, 18, 88. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Parrinello, N.L.; La Cava, P.; Di Rosa, M.; Bramanti, V.; Di Raimondo, C.; Conticello, C.; Chiarenza, A.; Palumbo, G.A.; et al. Granulocyte-like myeloid derived suppressor cells (G-MDSC) are increased in multiple myeloma and are driven by dysfunctional mesenchymal stem cells (MSC). Oncotarget 2016, 7, 85764–85775. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Parrinello, N.L.; La Cava, P.; Tibullo, D.; Giallongo, C.; Camiolo, G.; Puglisi, G.; Parisi, M.; Pirosa, M.C.; Martin, E.; et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert Rev. Mol. Diagn. 2018, 18, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, X.; Liu, H.; Zhao, P.; Chen, Y.; Luo, Y.; Zhang, Z.; Wang, X. Mesenchymal stromal cells enhance the suppressive effects ofmyeloid-derived suppressor cells of multiple myeloma. Leuk. Lymphoma 2017, 58, 2668–2676. [Google Scholar] [CrossRef]
- Van Valckenborgh, E.; Schouppe, E.; Movahedi, K.; De Bruyne, E.; Menu, E.; De Baetselier, P.; Vanderkerken, K.; Van Ginderachter, J.A. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia 2012, 26, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Shen, Q.; Lin, H.; Hu, L.; Li, G.; Zhang, X. Decitabine shows potent antimyeloma activity by depleting monocytic myeloid-derived suppressor cells in the myeloma microenvironment. J. Cancer Res. Clin. Oncol. 2019, 145, 329–336. [Google Scholar] [CrossRef]
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chrétien, M.L.; Guillerey, C.; Putz, E.M.; Bald, T.; Förster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 is a key driver of immunosuppression and a possible therapeutic target in the multiple myeloma microenvironment. Cancer Cell 2018, 33, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloidderived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.F.; Chen, Y.Y.; He, Y.Y.; Wang, J.Y.; Yang, J.P.; Zhong, S.L.; Jiang, N.; Zhou, P.; Jiang, J.; Zhou, J. Expansion and activation of granulocytic, myeloid-derived suppressor cells in childhood precursor B cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2017, 102, 449–458. [Google Scholar] [CrossRef]
- Wang, L.; Jia, B.; Claxton, D.F.; Christopher Ehmann, W.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology 2018, 7, e1469594. [Google Scholar] [CrossRef]
- Hohtari, H.; Bruck, O.; Blom, S.; Turkki, R.; Sinisalo, M.; Kovanen, P.E.; Kallioniemi, O.; Pellinen, T.; Porkka, K.; Mustjoki, S. Immune cell constitution in bone marrow microenvironment predicts outcome in adult ALL. Leukemia 2019, 33, 1570–1582. [Google Scholar] [CrossRef] [Green Version]
- Trabanelli, S.; Chevalier, M.F.; Martinez-Usatorre, A.; Gomez-Cadena, A.; Salome, B.; Lecciso, M.; Salvestrini, V.; Veldeil, G.; Racle, J.; Papayannidis, C.; et al. Tumourderived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun. 2017, 8, 593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansson, L.; Soderlund, S.; Mangsbo, S.; Hjorth-Hansen, H.; Höglund, M.; Markevärn, B.; Richter, J.; Stenke, L.; Mustjoki, S.; Loskong, A.; et al. The tyrosine kinase inhibitors imatinib and dasatinib reduce myeloid suppressor cells and release effector lymphocyte responses. Mol. Cancer Ther. 2015, 14, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallongo, C.; Parrinello, N.; Tibullo, D.; La Cava, P.; Romano, A.; Chiarenza, A.; Barbagallo, I.; Palumbo, G.A.; Stagno, F.; Vigneri, P.; et al. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS ONE 2014, 9, e101848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallongo, C.; Parrinello, N.L.; La Cava, P.; Camiolo, G.; Romano, A.; Scalia, M.; Stagno, F.; Palumbo, G.A.; Avola, R.; Volti, G.L.; et al. Monocytic myeloid-derived suppressor cells as prognostic factor in chronic myeloid leukaemia patients treated with dasatinib. J. Cell Mol. Med. 2018, 22, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Alves, R.; McArdle, S.E.; Vadakekolathu, J.; Gonçalves, A.C.; Freitas-Tavares, P.; Pereira, A.; Almedia, A.M.; Sarmento-Ribeiro, A.B.; Rutella, S. Flow cytometry and targeted immune transcriptomics identify distinct profiles in patients with chronic myeloid leukemia receiving tyrosine kinase inhibitors with or without interferon-alpha. J. Transl. Med. 2020, 18, 2. [Google Scholar] [CrossRef]
- Taleb, K.; Auffray, C.; Villefroy, P.; Pereira, A.; Hosmalin, A.; Gaudry, M.; Le Bon, A. Chronic type I IFN is sufficient to promote immunosuppression through accumulation of myeloid-derived suppressor cells. J. Immunol. 2017, 198, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLADRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhou, Y.; Huang, Q.; Qiu, L. CD14(+) HLA-DR(low/−) expression: A novel prognostic factor in chronic lymphocytic leukemia. Oncol. Lett. 2015, 9, 1167–1172. [Google Scholar] [CrossRef] [Green Version]
- Zahran, A.M.; Moeen, S.M.; Thabet, A.F.; Rayan, A.; Abdel-Rahim, M.H.; Mohamed, W.M.Y.; Hetta, H.F. Monocytic myeloid-derived suppressor cells in chronic lymphocytic leukemia patients: A single center experience. Leuk. Lymphoma 2020, 61, 1645–1652. [Google Scholar] [CrossRef]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef]
- Duan, J.; Wang, Y.; Jiao, S. Checkpoint blockade-based immunotherapy in the context of tumor microenvironment: Opportunities and challenges. Cancer Med. 2018, 7, 4517–4529. [Google Scholar] [CrossRef] [Green Version]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G. Cancer Immunotherapy and Breaking Immune Tolerance-New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Ghobrial, I.M. Immunotherapy for hematological malignancies. J. Life Sci. 2019, 1, 46–52. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar]
- Dhodapkar, M.V.; Dhodapkar, K.M. Immune modulation in hematologic malignancies. Semin. Oncol. 2015, 42, 617–625. [Google Scholar] [CrossRef] [Green Version]
- Forte, D.; Krause, D.S.; Andreeff, M.; Bonnet, D.; Méndez-Ferrer, S. Updates on the hematologic tumor microenvironment and its therapeutic targeting. Haematologica 2019, 104, 1928–1934. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer. 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Bachireddy, P.; Burkhardt, U.E.; Rajasagi, M.; Wu, C.J. Hematologic malignancies: At the forefront of immunotherapeutic innovation. Nat. Rev. Cancer 2015, 15, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef]
- Pianko, M.J.; Goldberg, A.D.; Lesokhin, A.M. Clinical Development of PD-1 Blockade in Hematologic Malignancies. Cancer J. 2018, 24, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Annibali, O.; Crescenzi, A.; Tomarchio, V.; Pagano, A.; Bianchi, A.; Grifoni, A.; Avvisati, G. PD-1/PD-L1 checkpoint in hematological malignancies. Leuk. Res. 2018, 67, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.M. A Nobel Prize-worthy pursuit: Cancer immunology and harnessing immunity to tumour neoantigens. Immunology 2018, 155, 283–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernadic, M., Jr.; Duchon, R.; Aziri, R.; Mladosievicova, B. New principles of cancer therapy give new hope for oncological patients. Bratisl. Lek. Listy 2019, 120, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.Y.; Ahmed, R.; Okazaki, T. Role of PD-1 in regulating T-cell immunity. Curr. Top. Microbiol. Immunol. 2011, 350, 17–37. [Google Scholar]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2019, 229, 114–125. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, L.; Li, J.; Li, Y.; Wang, Y.; Xu, Z.X. Recent Findings in the Regulation of Programmed Death Ligand 1 Expression. Front. Immunol. 2019, 10, 1337. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, G.; Lee, Y.C.; Luh, F.; Kuo, C.N.; Chang, W.C.; Yen, Y. Development and clinical applications of cancer immunotherapy against PD-1 signaling pathway. J. Biomed. Sci. 2019, 26, 96. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Mar, S.; Donoso-Quezada, J.; González-Valdez, J. Clinical Implications of Exosomal PD-L1 in Cancer Immunotherapy. J. Immunol. Res. 2021, 8, 8839978. [Google Scholar]
- Ye, Q.; Wang, C.; Xian, J.; Zhang, M.; Cao, Y.; Cao, Y. Expression of programmed cell death protein 1 (PD-1) and indoleamine 2,3-dioxygenase (IDO) in the tumor microenvironment and in tumor-draining lymph nodes of breast cancer. Hum. Pathol. 2018, 75, 81–90. [Google Scholar] [CrossRef]
- Patil, P.A.; Blakely, A.M.; Lombardo, K.A.; Machan, J.T.; Miner, T.J.; Wang, L.J.; Marwaha, A.S.; Matoso, A. Expression of PD-L1, indoleamine 2,3-dioxygenase and the immune microenvironment in gastric adenocarcinoma. Histopathology 2018, 73, 124–136. [Google Scholar] [CrossRef]
- Khoja, L.; Butler, M.O.; Kang, S.P.; Ebbinghaus, S.; Joshua, A.M. Pembrolizumab. J. Immunother. Cancer 2015, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Fulcheiro, E.; Jimeno, A. Nivolumab. Drugs Today 2014, 50, 791–802. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Zhou, J.; Young, K.H. PD-1 expression and clinical PD-1 blockade in B-cell lymphomas. Blood 2018, 131, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Muenst, S.; Hoeller, S.; Willi, N.; Dirnhofera, S.; Tzankov, A. Diagnostic and prognostic utility of PD-1 in B cell lymphomas. Dis. Markers 2010, 29, 47–53. [Google Scholar] [CrossRef]
- Gopas, J.; Stern, E.; Zurgil, U.; Ozer, J.; Ben-Ari, A.; Shubinsky, G.; Braiman, A.; Sinay, R.; Ezratty, J.; Dronov, V.; et al. Reed-Sternberg cells in Hodgkin’s lymphoma present features of cellular senescence. Cell Death Dis. 2016, 7, e2457. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wenzl, K.; Manske, M.K.; Asmann, Y.W.; Sarangi, V.; Greipp, P.; Krull, J.E.; Hartert, K.; He, R.; Feldman, A.L.; et al. Amplification of 9p24.1 in diffuse large B-cell lymphoma identifies a unique subset of cases that resemble primary mediastinal large B-cell lymphoma. Blood Cancer J. 2019, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Barrett, M.T.; Anderson, K.S.; Lenkiewicz, E.; Andreozzi, M.; Cunliffe, H.E.; Klassen, C.L.; Dueck, A.C.; McCullough, A.E.; Reddy, S.K.; Ramanathan, R.K.; et al. Genomic amplification of 9p24.1 targeting JAK2, PD-L1, and PD-L2 is enriched in high-risk triple negative breast cancer. Oncotarget 2015, 6, 26483–26493. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Armand, P.; Timmerman, J.M.; Shipp, M.A.; Garelik, M.B.B.; Zhu, L. Nivolumab in Patients (Pts) with Relapsed or Refractory Classical Hodgkin Lymphoma (R/R cHL): Clinical Outcomes from Extended Follow-up of a Phase 1 Study (CA209-039). Blood 2015, 126. [Google Scholar] [CrossRef]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S.; et al. Programmed Death-1 Blockade With Pembrolizumab in Patients With Classical Hodgkin Lymphoma After Brentuximab Vedotin Failure. J. Clin. Oncol. 2016, 34, 3733–3739. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Manson, G.; Mear, J.-B.; Herbaux, C.; Schiano, J.-M.; Casasnovas, O.; Stamatoullas, A.; Deau, B.; Schmitt, A.; Garnier, G.; Regny, C.; et al. Long-term efficacy of anti-PD1 therapy in Hodgkin lymphoma with and without allogenic stem cell transplantation. Eur. J. Cancer 2019, 115, 47–56. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Ribrag, V.; Moskowitz, C.H.; Michot, J.M.; Kuruvilla, J.; Balakumaran, A.; Zhang, Y.; Chlosta, S.; Shipp, M.A.; Armand, P. Safety and tolerability of pembrolizumab in patients with relapsed/refractory primary mediastinal large B-cell lymphoma. Blood 2017, 130, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Nayak, L.; Iwamoto, F.M.; LeCasce, A.; Mukundan, S.; Roemer, M.G.M.; Chapuy, B.; Armand, P.; Rodig, S.J.; Shipp, M.A. PD-1 blockade with nivolumab in relapsed/refractory primary central nervous system and testicular lymphoma. Blood 2017, 129, 3071–3073. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Ricken, G.; Widhalm, G.; Rajky, O.; Hainfellner, J.A.; Birner, P.; Raderer, M.; Preusser, M. PD1 (CD279) and PD-L1 (CD274, B7H1) expression in primary central nervous system lymphomas (PCNSL). Clin. Neuropathol. 2014, 33, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Jung, H.Y.; Nam, S.J.; Kim, T.M.; Heo, D.S.; Kim, C.W.; Jeon, Y.K. Expression of programmed cell death ligand 1 (PD-L1) in advanced stage EBV-associated extranodal NK/T cell lymphoma is associated with better prognosis. Virchows Arch. 2016, 469, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Kiyasu, J.; Miyoshi, H.; Hirata, A.; Arakawa, F.; Ichikawa, A.; Niino, D.; Sugita, Y.; Yufu, Y.; Choi, I.; Abe, Y.; et al. Expression of programmed cell death ligand 1 is associated with poor overall survival in patients with diffuse large B-cell lymphoma. Blood 2015, 126, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Rossille, D.; Gressier, M.; Damotte, D.; Maucort-Boulch, D.; Pangault, C.; Semana, G.; Le Gouill, S.; Haioun, C.; Tarte, K.; Lamy, L.; et al. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: Results from a French multicenter clinical trial. Leukemia 2014, 28, 2367–2375. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.; Asmann, Y.; Cattaneo, L.; Braggio, E.; Keats, J.; Auclair, D.; Lonial, S.; Russell, S.J.; Stewart, A.K. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017, 7, e612. [Google Scholar] [CrossRef] [Green Version]
- Ocio, E.M.; Mateos, M.V.; Orlowski, R.Z.; Siegel, D.; Reece, D.E.; Moreau, P.; Munshi, N.; Avigan, D.E.; Siegel, D.S.; Ghori, R.; et al. Pembrolizumab (Pembro) plus lenalidomide (Len) and low-dose dexamethasone (Dex) for relapsed/refractory multiple myeloma (RRMM): Efficacy and biomarker analyses. J. Clin. Oncol. 2017, 35, 8015. [Google Scholar] [CrossRef]
- Badros, A.; Hyjek, E.; Ma, N.; Lesokhin, A.; Dogan, A.; Rapoport, A.P.; Kocoglu, M.; Lederer, E.; Philip, S.; Milliron, T.; et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood 2017, 130, 1189–1197. [Google Scholar] [CrossRef] [Green Version]
- Mateos, M.-V.; Blacklock, H.; Schjesvold, F.; Orial, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomized, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Administration FaD. Keytruda (Pembrolizumab) in Patients with Multiple Myeloma: FDA Statement—Two Clinical Trials on Hold; U.S. Food and Drug Administration: Washington, DC, USA, 2017.
- Administration USFD. FDA Alerts Healthcare Professionals and Oncology Clinical Investigators about Two Clinical Trials on Hold Evaluating KEYTRUDA® (Pembrolizumab) in Patients with Multiple Myeloma; U.S. Food and Drug Administration: Washington, DC, USA, 2017.
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Chen, X.; Liu, S.; Wang, L.; Zhang, W.; Ji, Y.; Ma, X. Clinical significance of B7-H1 (PD-L1) expression in human acute leukemia. Cancer Biol. Ther. 2008, 7, 622–627. [Google Scholar] [CrossRef] [Green Version]
- Salih, H.R.; Wintterle, S.; Krusch, M.; Kroner, A.; Huang, Y.H.; Chen, L.; Wiendl, H. The role of leukemia-derived B7-H1 (PD-L1) in tumor-T-cell interactions in humans. Exp. Hematol. 2006, 34, 888–894. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Lichtenegger, F.S.; Köhnke, T.; Vick, B.; Jeremias, I.; Metzeler, K.H.; Altmann, T.; et al. Blockade of the PD-1/PD-L1 axis augments lysis of AML cells by the CD33/CD3 BiTE antibody construct AMG 330: Reversing a T-cell-induced immune escape mechanism. Leukemia 2016, 30, 484–491. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Daver, N.G.; Montalban-Bravo, G.; Jabbour, E.J.; DiNardo, C.D.; Kornblau, S.M.; Bose, P.; Alvarado, Y.; Ohanian, M.; Borthakur, G.; et al. A Phase II Study Evaluating the Combination of Nivolumab (Nivo) or Ipilimumab (Ipi) with Azacitidine in Pts with Previously Treated or Untreated Myelodysplastic Syndromes (MDS). Blood 2016, 128, 344. [Google Scholar] [CrossRef]
- Cassaday, R.D.; Garcia, K.L.A.; Fromm, J.R.; Percival, M.E.M.; Turtle, C.J.; Nghiem, P.T.; Stevenson, P.A.; Estey, E.H. Phase 2 study of pembrolizumab for measurable residual disease in adults with acute lymphoblastic leukemia. Blood Adv. 2020, 4, 3239–3245. [Google Scholar] [CrossRef]
- Barta, S.K.; Zain, J.; MacFarlane, A.W.; Smith, S.M.; Ruan, J.; Fung, H.C.; Tan, C.R.; Yang, Y.; Alpaugh, R.K.; Dulami, E.; et al. Phase II Study of the PD-1 Inhibitor Pembrolizumab for the Treatment of Relapsed or Refractory Mature T-cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 356–364. [Google Scholar] [CrossRef]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells—a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Haile, L.A.; Greten, T.F.; Korangy, F. Immune suppression: The hallmark of myeloid derived suppressor cells. Immunol. Investig. 2012, 41, 581–594. [Google Scholar] [CrossRef]
- Bian, Z.; Abdelaal, A.M.; Shi, L.; Liang, H.; Xiong, L.; Kidder, K.; Venkataramani, M.; Culpepper, C.; Zen, K.; Liu, Y. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T-cell proliferation. Eur. J. Immunol. 2018, 48, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Cané, S.; Bronte, V. Detection and functional evaluation of arginase-1 isolated from human PMNs and murine MDSC. Methods Enzymol. 2020, 632, 193–213. [Google Scholar]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [Green Version]
- Ballbach, M.; Dannert, A.; Singh, A.; Siegmund, D.M.; Handgretinger, R.; Piali, L.; Rieber, N.; Hartl, D. Expression of checkpoint molecules on myeloid-derived suppressor cells. Immunol. Lett. 2017, 192, 1–6. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef]
- Condamine, T.; Ramachandran, I.; Youn, J.I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoskioka, Y.; Koshiyama, M.; Konishi, I. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilatova, K.; Bencsikova, B.; Demlova, R.; Valik, D.; Zdrazilova-Dubska, L. Myeloid-derived suppressor cells (MDSCs) in patients with solid tumors: Considerations for granulocyte colony-stimulating factor treatment. Cancer Immunol. Immunother. 2018, 67, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 783. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef] [Green Version]
- Gravelle, P.; Burroni, B.; Péricart, S.; Rossi, C.; Bezombes, C.; Tosolini, M.; Damotte, D.; Brousset, P.; Fournié, J.J.; Laurent, C. Mechanisms of PD-1/PD-L1 expression and prognostic relevance in non-Hodgkin lymphoma: A summary of immunohistochemical studies. Oncotarget 2017, 8, 44960–44975. [Google Scholar] [CrossRef] [Green Version]
- Azzaoui, I.; Uhel, F.; Rossille, D.; Pangault, C.; Dulong, J.; Le Priol, J.; Lamy, T.; Houot, R.; Le Gouill, S.; Cartron, G.; et al. T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 2016, 128, 1081–1092. [Google Scholar] [CrossRef]
- Christiansson, L.; Söderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S.I. Increased Level of Myeloid-Derived Suppressor Cells, Programmed Death Receptor Ligand 1/Programmed Death Receptor 1, and Soluble CD25 in Sokal High Risk Chronic Myeloid Leukemia. PLoS ONE 2013, 8, e55818. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, Y.; Nonomura, C.; Kondou, R.; Miyata, H.; Ashizawa, T.; Maeda, C.; Sugino, T.; Yamaguichi, K.; Ando, T.; Ishikawa, Y.; et al. Immunological effects of the anti-programmed death-1 antibody on human peripheral blood mononuclear cells. Int. J. Oncol. 2015, 49, 1099–1107. [Google Scholar] [CrossRef] [Green Version]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment, phenotype, properties, and mechanisms of immune suppression. Semin. Cancer Biol. 2006, 16, 53–65. [Google Scholar] [CrossRef]
- Tallón de Lara, P.; Cecconi, V.; Hiltbrunner, S.; Yagita, H.; Friess, M.; Bode, B.; Optiz, I.; Vrugt, B.; Weder, W.; Stolzmann, P.; et al. Gemcitabine Synergizes with Immune Checkpoint Inhibitors and Overcomes Resistance in a Preclinical Model and Mesothelioma Patients. Clin. Cancer Res. 2018, 24, 6345–6354. [Google Scholar] [CrossRef] [Green Version]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, S. Immunogenic Chemotherapy Sensitizes Renal Cancer to Immune Checkpoint Blockade Therapy in Preclinical Models. Med. Sci. Monit. 2017, 23, 3360–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.T.; Mao, L.; Wu, L.; Deng, W.W.; Bu, L.L.; Liu, J.F.; Chen, L.; Yang, L.L.; Wu, Y.; Zhang, W.F.; et al. Inhibition of SRC family kinases facilitates anti-CTLA4 immunotherapy in head and neck squamous cell carcinoma. Cell Mol. Life Sci. 2018, 75, 4223–4234. [Google Scholar] [CrossRef]
- Westerweel, P.E.; te Boekhorst, P.A.W.; Levin, M.D.; Cornelissen, J.J. New Approaches and Treatment Combinations for the Management of Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 665. [Google Scholar] [CrossRef]
- Moura, J.; Rodrigues, J.; Santos, A.H.; Teixeira, M.A.; Queiros, M.L.; Santos, M.; Gonçalves, M.; Fonseca, S.; Laranjeira, C.; Rodrigues, A.S.; et al. Chemokine receptor repertoire reflects mature T-cell lymphoproliferative disorder clinical presentation. Blood Cells Mol. Dis. 2009, 42, 57–63. [Google Scholar] [CrossRef]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [Green Version]
- Briere, D.; Sudhakar, N.; Woods, D.M.; Hallin, J.; Engstrom, L.D.; Aranda, R.; Chiang, H.; Sodré, A.L.; Olson, P.; Weber, J.S.; et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol. Immunother. 2018, 67, 381–392. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- Stiff, A.; Trikha, P.; Weselowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Barsoumian, H.B.; Schoenhals, J.E.; Cushman, T.R.; Caetano, M.S.; Wang, X.; Valdecanas, D.R.; Nikman, S.; Younes, A.I.; Li, G.; et al. Indoleamine 2,3-dioxygenase 1 inhibition targets anti-PD1-resistant lung tumors by blocking myeloid-derived suppressor cells. Cancer Lett. 2018, 431, 54–63. [Google Scholar] [CrossRef]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [Green Version]
- Iclozan, C.; Antonia, S.; Chiappori, A.; Chen, D.T.; Gabrilovich, D.I. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. 2013, 62, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Tobin, R.P.; Jordan, K.R.; Robinson, W.A.; Davis, D.; Borges, V.F.; Gonzalez, R.; Lewis, K.D.; McCarter, M.D. Targeting myeloid-derived suppressor cells using all-trans retinoic acid in melanoma patients treated with Ipilimumab. Int. Immunopharmacol. 2018, 63, 282–291. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Passaro, A.; Mansuco, P.; Gandini, S.; Spitaleri, G.; Labanca, V.; Guerini-Rocco, E.; Barberis, M.; Catania, C.; Del Signore, E.; de Marinis, F.; et al. Gr-MDSC-linked asset as a potential immune biomarker in pretreated NSCLC receiving nivolumab as second-line therapy. Clin. Transl. Oncol. 2020, 22, 603–611. [Google Scholar] [CrossRef]
- Fedorova, L.; Pilatova, K.; Selingerova, I.; Bencsikova, B.; Budinska, E.; Zwinsova, B.; Brychtová, V.; Langrová, M.; Sefr, R.; Valík, D.; et al. Circulating Myeloid-Derived Suppressor Cell Subsets in Patients with Colorectal Cancer—Exploratory Analysis of Their Biomarker Potential. Klin. Onkol. 2018, 31 (Suppl. S2), 88–92. [Google Scholar] [CrossRef]
- Gunes, E.G.; Rosen, S.T.; Querfeld, C. The role of myeloid-derived suppressor cells in hematologic malignancies. Curr. Opin. Oncol. 2020, 32, 518–526. [Google Scholar] [CrossRef]
- Betsch, A.; Rutgeerts, O.; Fevery, S.; Sprangers, B.; Verhoef, G.; Dierickx, D.; Beckers, M. Myeloid-derived suppressor cells in lymphoma: The good, the bad and the ugly. Blood Rev. 2018, 32, 490–498. [Google Scholar] [CrossRef]
- Lewinsky, H.; Gunes, E.G.; David, K.; Radomir, L.; Kramer, M.P.; Pellegrino, B.; Perpinial, M.; Chen, J.; He, T.F.; Mansour, A.G.; et al. CD84 is a regulator of the immunosuppressive microenvironment in multiple myeloma. JCI Insight 2021, 6, e141683. [Google Scholar]
- D’Aveni, M.; Notarantonio, A.B.; Bertrand, A.; Boulangé, L.; Pochon, C.; Rubio, M.T. Myeloid-Derived Suppressor Cells in the Context of Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2020, 11, 989. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, H.M.; Ma, G.; Zhou, Z.; Raulet, D.; Rivera, A.L.; Chen, S.H.; Pan, P.-Y. The mechanistic study behind suppression of GVHD while retaining GVL activities by myeloid-derived suppressor cells. Leukemia 2019, 33, 2078–2089. [Google Scholar] [CrossRef]

{kind=link}
| MDSC Population | Chemokine Receptor | Cluster of Differentiation * | MHC | Ly6 | Mac-2 (Galectin-3) |
|---|---|---|---|---|---|
| M-MDSC | CCR2 (high) CCR5 CX3CR1 CXCR1 CXCR2 CXCR4 | CD1b CD49d CD54 (high) CD71 CD73 | Class I Class II (+/−) | Ly6C + (high) | High |
| PMN-MDSC | CCR2 CCR5 CX3CR1 CXCR1 CXCR2 CXCR4 | CD43 CD54 (low) CD73 (high) CD98 CD244 (+/−) | Class I Class II (+/−) | Ly6C + (low) Ly6G + (high) | Low |
| Type of MDSC | Chemokine Receptor | Cluster of Differentiation * | HLA-DR | Lin |
|---|---|---|---|---|
| M-MDSC | CCR2 (high) CXCR4 CXCR1 CXCR2 | CD14 (high) CD68 CD80 (+/−) CD83 (+/−) CD86 (+/−) CD163 CD117 (+/−) | Negative | +/− (low) |
| PMN-MDSC | CCR2 CXCR4 CXCR1 CXCR2 | CD15 CD66b CD117 | Negative | +/− (low) |
| Haematological Malignancies | NCT Number | Interventions |
|---|---|---|
| CML, BCR-ABL1-positive minimal residual disease | NCT03516279 | Pembrolizumab and dasatinib, imatinib mesylate, or nilotinib in treating patients with CML and persistently detectable minimal residual disease |
| Myelodysplastic syndrome | NCT02936752 | Entinostat and pembrolizumab in treating patients with myelodysplastic syndrome after DNMTi therapy failure |
| Recurrent CCL | NCT02332980 | Pembrolizumab alone or with idelalisib or ibrutinib in treating patients with R/R chronic lymphocytic leukaemia or other low-grade B cell NHL |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivares-Hernández, A.; Figuero-Pérez, L.; Terán-Brage, E.; López-Gutiérrez, Á.; Velasco, Á.T.; Sarmiento, R.G.; Cruz-Hernández, J.J.; Miramontes-González, J.P. Resistance to Immune Checkpoint Inhibitors Secondary to Myeloid-Derived Suppressor Cells: A New Therapeutic Targeting of Haematological Malignancies. J. Clin. Med. 2021, 10, 1919. https://doi.org/10.3390/jcm10091919
Olivares-Hernández A, Figuero-Pérez L, Terán-Brage E, López-Gutiérrez Á, Velasco ÁT, Sarmiento RG, Cruz-Hernández JJ, Miramontes-González JP. Resistance to Immune Checkpoint Inhibitors Secondary to Myeloid-Derived Suppressor Cells: A New Therapeutic Targeting of Haematological Malignancies. Journal of Clinical Medicine. 2021; 10(9):1919. https://doi.org/10.3390/jcm10091919
Chicago/Turabian StyleOlivares-Hernández, Alejandro, Luis Figuero-Pérez, Eduardo Terán-Brage, Álvaro López-Gutiérrez, Álvaro Tamayo Velasco, Rogelio González Sarmiento, Juan Jesús Cruz-Hernández, and José Pablo Miramontes-González. 2021. "Resistance to Immune Checkpoint Inhibitors Secondary to Myeloid-Derived Suppressor Cells: A New Therapeutic Targeting of Haematological Malignancies" Journal of Clinical Medicine 10, no. 9: 1919. https://doi.org/10.3390/jcm10091919