
Modulation of Bile Acid Metabolism to Improve Plasma Lipid and Lipoprotein Profiles


Abstract
:1. Introduction
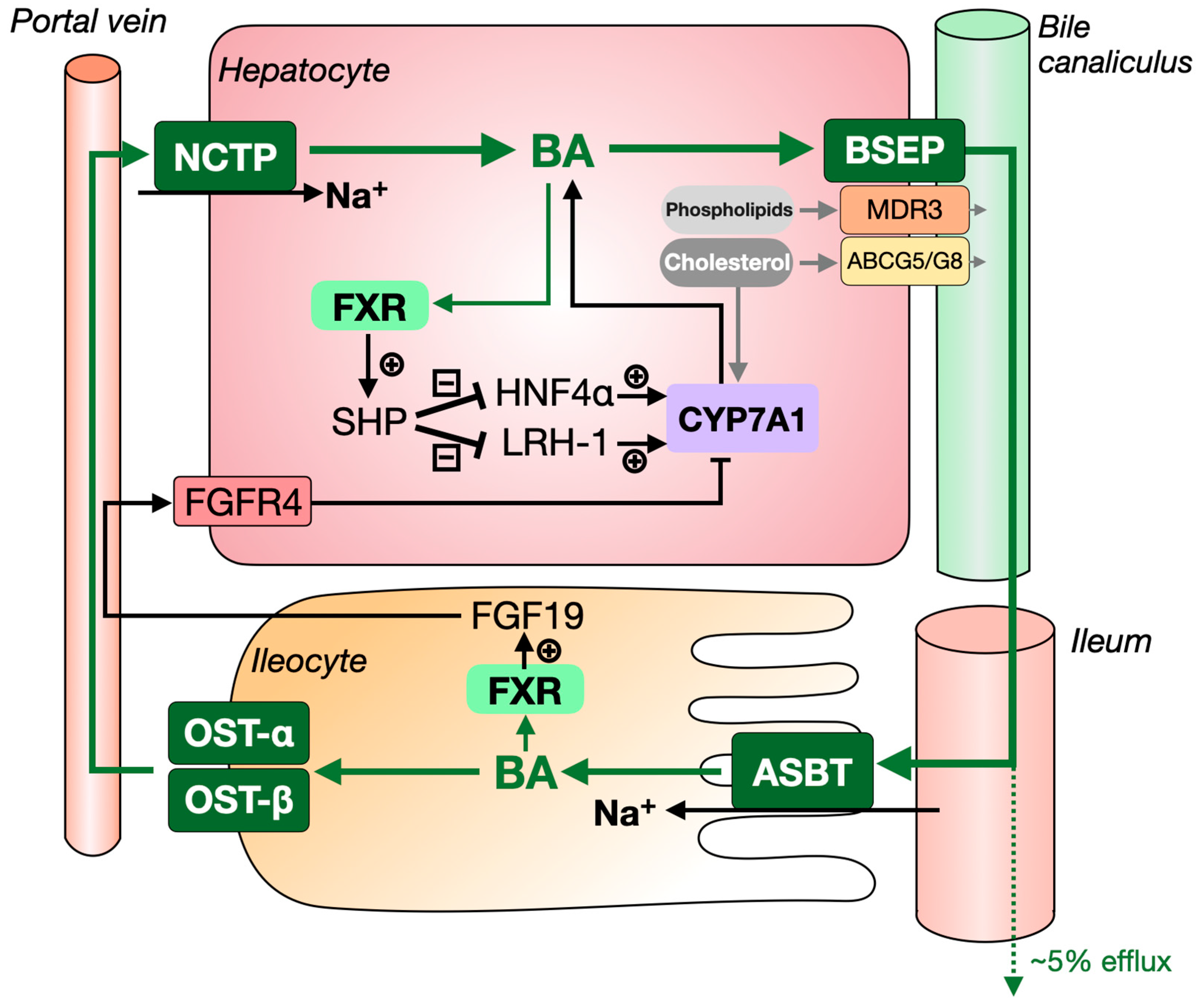
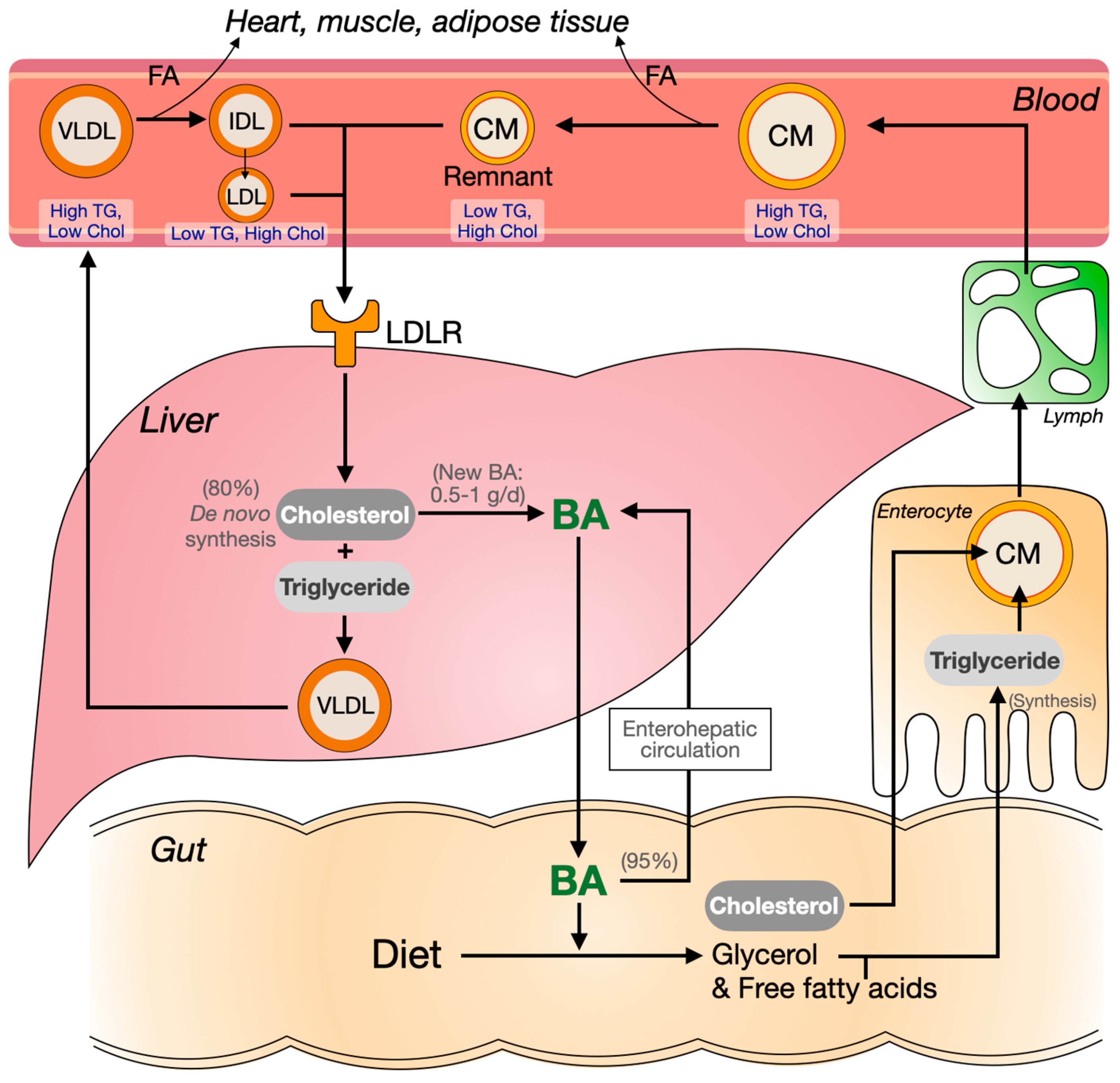
1.1. The Synthesis and Enterohepatic Circulation of Bile Acids
1.2. Bile Formation and Its Role in Intestinal Lipid Absorption
1.3. Bile Acids as Signalling Molecules
1.4. Cholesterol, Triglycerides and Plasma Lipoprotein Metabolism
1.5. Challenges and Aim of This Review
2. Intervening in the Enterohepatic Circulation of Bile Acids
2.1. Bile Acid Sequestrants
2.1.1. Mechanism of Action
2.1.2. BAS as Monotherapy
- Treating familial hypercholesterolemia (FH)
- Treating type 2 diabetes (T2D)
2.1.3. Bile Acid Sequestrants Combined with Other Drugs
- Treating primary dyslipidemia
- Treating type 2 diabetes
- Combinations of BAS with ezetimibe and statins
2.2. ASBT Inhibitors
2.2.1. Elobixibat (A3309)
2.2.2. Linerixibat and Odevixibat
2.3. Bile-Salt Export Pump (BSEP), Organic Solute Transporter-α/β (OST-α/β) and Sodium Dependent Taurocholate Cotransport Peptide (NTCP)
3. Bile Acid Synthesis and Plasma Lipids
3.1. FXR and TGR5 Agonists
3.1.1. FXR Agonists
3.1.2. TGR5 Agonists
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Charach, G.; Rabinovich, P.D.; Konikoff, F.M.; Grosskopf, I.; Weintraub, M.S.; Gilat, T. Decreased fecal bile acid output in patients with coronary atherosclerosis. J. Med. 1998, 29, 125–136. [Google Scholar]
- Charach, G.; Argov, O.; Geiger, K.; Charach, L.; Rogowski, O.; Grosskopf, I. Diminished bile acids excretion is a risk factor for coronary artery disease: 20-year follow up and long-term outcome. Therap. Adv. Gastroenterol. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charach, G.; Karniel, E.; Novikov, I.; Galin, L.; Vons, S.; Grosskopf, I.; Charach, L. Reduced bile acid excretion is an independent risk factor for stroke and mortality: A prospective follow-up study. Atherosclerosis 2020, 293, 79–85. [Google Scholar] [CrossRef]
- The Lipid Research Clinics Coronary Primary Prevention Trial Results: I. Reduction in Incidence of Coronary Heart Disease. JAMA J. Am. Med. Assoc. 1984, 251, 351–364. [CrossRef]
- The Lipid Research Clinics Coronary Primary Prevention Trial Results: II. The Relationship of Reduction in Incidence of Coronary Heart Disease to Cholesterol Lowering. JAMA J. Am. Med. Assoc. 1984, 251, 365–374. [CrossRef]
- Qayyum, F.; Lauridsen, B.K.; Frikke-Schmidt, R.; Kofoed, K.F.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Genetic variants in CYP7A1 and risk of myocardial infarction and symptomatic gallstone disease. Eur. Heart J. 2018, 39, 2106–2116. [Google Scholar] [CrossRef]
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y.L. Regulation of bile acid synthesis: Pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004, 40, 539–551. [Google Scholar] [CrossRef]
- Chen, L.; van den Munckhof, I.C.L.; Schraa, K.; ter Horst, R.; Koehorst, M.; van Faassen, M.; van der Ley, C.; Doestzada, M.; Zhernakova, D.V.; Kurilshikov, A.; et al. Genetic and Microbial Associations to Plasma and Fecal Bile Acids in Obesity Relate to Plasma Lipids and Liver Fat Content. Cell Rep. 2020, 33, 108–212. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, V.I. Physiological and molecular biochemical mechanisms of bile formation. World J. Gastroenterol. 2013, 19, 7341–7360. [Google Scholar] [CrossRef]
- De Boer, J.F.; Bloks, V.W.; Verkade, E.; Heiner-Fokkema, M.R.; Kuipers, F. New insights in the multiple roles of bile acids and their signaling pathways in metabolic control. Curr. Opin. Lipidol. 2018, 29, 194–202. [Google Scholar] [CrossRef]
- Chiang, J.Y.L. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef] [Green Version]
- Strazzabosco, M. New insights into cholangiocyte physiology. J. Hepatol. 1997, 27, 945–952. [Google Scholar] [CrossRef]
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayagam, J.S.; Williamson, C.; Joshi, D.; Thompson, R.J. Review article: Liver disease in adults with variants in the cholestasis-related genes ABCB11, ABCB4 and ATP8B1. Aliment. Pharmacol. Ther. 2020, 52, 1628–1639. [Google Scholar] [CrossRef]
- Williams, K.; Segard, A.; Graf, G.A. Sitosterolemia: Twenty Years of Discovery of the Function of ABCG5ABCG8. Int. J. Mol. Sci. 2021, 22, 2641. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Terada, T. Molecular mechanisms for biliary phospholipid and drug efflux mediated by ABCB4 and bile salts. Biomed. Res. Int. 2014, 2014, 954781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticho, A.L.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Intestinal Absorption of Bile Acids in Health and Disease. Compr. Physiol. 2019, 10, 21–56. [Google Scholar] [CrossRef]
- Minich, D.M.; Havinga, R.; Stellaard, F.; Vonk, R.J.; Kuipers, F.; Verkade, H.J. Intestinal absorption and postabsorptive metabolism of linoleic acid in rats with short-term bile duct ligation. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G1242–G1248. [Google Scholar] [CrossRef] [PubMed]
- Minich, D.M.; Kalivianakis, M.; Havinga, R.; van Goor, H.; Stellaard, F.; Vonk, R.J.; Kuipers, F.; Verkade, H.J. Bile diversion in rats leads to a decreased plasma concentration of linoleic acid which is not due to decreased net intestinal absorption of dietary linoleic acid. Biochim. Biophys. Acta 1999, 1438, 111–119. [Google Scholar] [CrossRef]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bite acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Bile Acids as Hormones: The FXR-FGF15/19 Pathway. Dig. Dis. 2015, 33, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaudo, S.; Dongiovanni, P.; Pihlajamäki, J.; Eslam, M.; Yki-Järvinen, H.; Pipitone, R.M.; Baselli, G.; Cammà, C.; Di Marco, V.; Enea, M.; et al. NR1H4 rs35724 G>C Variant Modulates Liver Damage in Nonalcoholic Fatty Liver Disease. Dig. Liver Dis. 2021, 53, S25. [Google Scholar] [CrossRef]
- Deaton, A.M.; Sulem, P.; Nioi, P.; Benonisdottir, S.; Ward, L.D.; Davidsson, O.B.; Lao, S.; Helgadottir, A.; Fan, F.; Jensson, B.O.; et al. A rare missense variant in NR1H4 associates with lower cholesterol levels. Commun. Biol. 2018, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Amar, M.J.A.; Guo, G.; Brewer, H.B.; Gonzalez, F.J.; Sinal, C.J. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J. Biol. Chem. 2003, 278, 2563–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Aguiar Vallim, T.Q.; Edwards, P.A. Bile acids have the gall to function as hormones. Cell Metab. 2009, 10, 162–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Zhou, E.; Hoeke, G.; Li, Z.; Eibergen, A.C.; Schonk, A.W.; Koehorst, M.; Boverhof, R.; Havinga, R.; Kuipers, F.; Coskun, T.; et al. Colesevelam enhances the beneficial effects of brown fat activation on hyperlipidaemia and atherosclerosis development. Cardiovasc. Res. 2020, 116, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neese, R.A.; Faix, D.; Kletke, C.; Wu, K.; Wang, A.C.; Shackleton, C.H.; Hellerstein, M.K. Measurement of endogenous synthesis of plasma cholesterol in rats and humans using MIDA. Am. J. Physiol. 1993, 264, E136–E147. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.F.; Kuipers, F.; Groen, A.K. Cholesterol Transport Revisited: A New Turbo Mechanism to Drive Cholesterol Excretion. Trends Endocrinol. Metab. 2018, 29, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Stellaard, F.; Brufau, G.; Boverhof, R.; Jonkers, E.Z.; Boer, T.; Kuipers, F. Developments in bile acid kinetic measurements using (13)C and (2)H: 10(5) times improved sensitivity during the last 40 years. Isotopes Environ. Health Stud. 2009, 45, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Stellaard, F.; Sackmann, M.; Berr, F.; Paumgartner, G. Simultaneous determination of pool sizes and fractional turnover rates, of deoxycholic acid, cholic acid and chenodeoxycholic acid in man by isotope dilution with 2H and 13C labels and serum sampling. Biomed. Environ. Mass Spectrom. 1987, 14, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Kuivenhoven, J.A.; Groen, A.K. Beyond the genetics of HDL: Why is HDL cholesterol inversely related to cardiovascular disease? Handb. Exp. Pharmacol. 2015, 224, 285–300. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Tybjærg-Hansen, A. Genetic determinants of LDL, lipoprotein(a), triglyceride-rich lipoproteins and HDL: Concordance and discordance with cardiovascular disease risk. Curr. Opin. Lipidol. 2011, 22, 113–122. [Google Scholar] [CrossRef]
- Vergeer, M.; Holleboom, A.G.; Kastelein, J.J.P.; Kuivenhoven, J.A. The HDL hypothesis: Does high-density lipoprotein protect from atherosclerosis? J. Lipid Res. 2010, 51, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Hovingh, G.K.; Rader, D.J.; Hegele, R.A. HDL re-examined. Curr. Opin. Lipidol. 2015, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B.; Bajaj, J.S. Bile acids and the gut microbiome. Curr. Opin. Gastroenterol. 2014, 30, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.M.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 2016, 57, 2130–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, J.F.; Verkade, E.; Mulder, N.L.; de Vries, H.D.; Huijkman, N.; Koehorst, M.; Boer, T.; Wolters, J.C.; Bloks, V.W.; van de Sluis, B.; et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J. Lipid Res. 2020, 61, 291–305. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Q.-H.; Tazuma, S.; Cohen, D.E.; Carey, M.C. Feeding natural hydrophilic bile acids inhibits intestinal cholesterol absorption: Studies in the gallstone-susceptible mouse. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G494–G502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef]
- Straniero, S.; Laskar, A.; Savva, C.; Härdfeldt, J.; Angelin, B.; Rudling, M. Of mice and men: Murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J. Lipid Res. 2020, 61, 480–491. [Google Scholar] [CrossRef] [Green Version]
- de Boer, J.F.; de Vries, H.D.; Palmiotti, A.; Li, R.; Doestzada, M.; Hoogerland, J.A.; Fu, J.; La Rose, A.M.; Westerterp, M.; Mulder, N.L.; et al. Cholangiopathy and Biliary Fibrosis in Cyp2c70-Deficient Mice Are Fully Reversed by Ursodeoxycholic Acid. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1045–1069. [Google Scholar] [CrossRef]
- De Vries, R.; Perton, F.G.; Dallinga-Thie, G.M.; van Roon, A.M.; Wolffenbuttel, B.H.R.; van Tol, A.; Dullaart, R.P.F. Plasma cholesteryl ester transfer is a determinant of intima-media thickness in type 2 diabetic and nondiabetic subjects: Role of CETP and triglycerides. Diabetes 2005, 54, 3554–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axelson, M.; Aly, A.; Sjövall, J. Levels of 7 alpha-hydroxy-4-cholesten-3-one in plasma reflect rates of bile acid synthesis in man. FEBS Lett. 1988, 239, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Axelson, M.; Björkhem, I.; Reihnér, E.; Einarsson, K. The plasma level of 7 alpha-hydroxy-4-cholesten-3-one reflects the activity of hepatic cholesterol 7 alpha-hydroxylase in man. FEBS Lett. 1991, 284, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Sjöberg, B.G.; Straniero, S.; Angelin, B.; Rudling, M. Cholestyramine treatment of healthy humans rapidly induces transient hypertriglyceridemia when treatment is initiated. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E167–E174. [Google Scholar] [CrossRef]
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staels, B.; Kuipers, F. Bile acid sequestrants and the treatment of type 2 diabetes mellitus. Drugs 2007, 67, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Blahová, T.; Peterková, L.; Leníček, M.; Vlachová, M.; Zemánková, K.; Adámková, V.; Vítek, L.; Kovář, J. The effect of colesevelam treatment on bile acid and lipid metabolism and glycemic control in healthy men. Physiol. Res. 2016, 65, 995. [Google Scholar] [CrossRef] [PubMed]
- Scaldaferri, F.; Pizzoferrato, M.; Ponziani, F.R.; Gasbarrini, G.; Gasbarrini, A. Use and indications of cholestyramine and bile acid sequestrants. Intern. Emerg. Med. 2013, 8, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M. The efficacy of colesevelam HCL in the treatment of heterozygous familial hypercholesterolemia in pediatric and adult patients. Clin. Ther. 2013, 35, 1247–1252. [Google Scholar] [CrossRef]
- Stein, E.A.; Marais, A.D.; Szamosi, T.; Raal, F.J.; Schurr, D.; Urbina, E.M.; Hopkins, P.N.; Karki, S.; Xu, J.; Misir, S.; et al. Colesevelam Hydrochloride: Efficacy and Safety in Pediatric Subjects with Heterozygous Familial Hypercholesterolemia. J. Pediatr. 2010, 156, 231–236.e3. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Pang, J.; Chan, D.C.; Brunt, J.N.; Lewis, B. Angiographic progression of coronary atherosclerosis in patients with familial hypercholesterolaemia treated with non-statin therapy: Impact of a fat-modified diet and a resin. Atherosclerosis 2016, 252, 82–87. [Google Scholar] [CrossRef]
- Kondo, K.; Kadowaki, T. Colestilan monotherapy significantly improves glycaemic control and LDL cholesterol levels in patients with type 2 diabetes: A randomized double-blind placebo-controlled study. Diabetes Obes. Metab. 2010, 12, 246–251. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Rosenson, R.S.; Hernandez-Triana, E.; Misir, S.; Jones, M.R. Colesevelam improved lipoprotein particle subclasses in patients with prediabetes and primary hyperlipidaemia. Diabetes Vasc. Dis. Res. 2013, 10, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Rigby, S.P.; Ford, D.M.; Tao, B.; Chou, H.S. The glucose and lipid effects of colesevelam as monotherapy in drug-naïve type 2 diabetes. Horm. Metab. Res. 2014, 46, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Angelin, B.; Hershon, K.S.; Brunzell, J.D. Bile acid metabolism in hereditary forms of hypertriglyceridemia: Evidence for an increased synthesis rate in monogenic familial hypertriglyceridemia. Proc. Natl. Acad. Sci. USA 1987, 84, 5434–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelin, B.; Einarsson, K.; Hellström, K.; Leijd, B. Bile acid kinetics in relation to endogenous tryglyceride metabolism in various types of hyperlipoproteinemia. J. Lipid Res. 1978, 19, 1004–1016. [Google Scholar] [CrossRef]
- Prawitt, J.; Abdelkarim, M.; Stroeve, J.H.M.; Popescu, I.; Duez, H.; Velagapudi, V.R.; Dumont, J.; Bouchaert, E.; Van Dijk, T.H.; Lucas, A.; et al. Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity. Diabetes 2011, 60, 1861–1871. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Bouchaert, E.; Abdelkarim, M.; Dumont, J.; Caron, S.; Fruchart, J.C.; Burcelin, R.; Kuipers, F.; Staels, B. FXR-deficiency confers increased susceptibility to torpor. FEBS Lett. 2007, 581, 5191–5198. [Google Scholar] [CrossRef]
- Claudel, T.; Inoue, Y.; Barbier, O.; Duran-Sandoval, D.; Kosykh, V.; Fruchart, J.; Fruchart, J.-C.; Gonzalez, F.J.; Staels, B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 2003, 125, 544–555. [Google Scholar] [CrossRef]
- Abel, U.; Schlüter, T.; Schulz, A.; Hambruch, E.; Steeneck, C.; Hornberger, M.; Hoffmann, T.; Perović-Ottstadt, S.; Kinzel, O.; Burnet, M.; et al. Synthesis and pharmacological validation of a novel series of non-steroidal FXR agonists. Bioorg. Med. Chem. Lett. 2010, 20, 4911–4917. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrema, H.; Meissner, M.; Dijk, T.H.V.; Brufau, G.; Boverhof, R.; Oosterveer, M.H.; Reijngoud, D.J.; Uller, M.M.; Stellaard, F.; Groen, A.K.; et al. Bile Salt Sequestration Induces Hepatic de Novo Lipogenesis Through Farnesoid X Receptor- and Liver X Receptorα-Controlled Metabolic Pathways in Mice. Hepatology 2010, 51, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Brufau, G.; Stellaard, F.; Prado, K.; Bloks, V.W.; Jonkers, E.; Boverhof, R.; Kuipers, F.; Murphy, E.J. Improved glycemic control with colesevelam treatment in patients with type 2 diabetes is not directly associated with changes in bile acid metabolism. Hepatology 2010, 52, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Beysen, C.; Murphy, E.J.; Deines, K.; Chan, M.; Tsang, E.; Glass, A.; Turner, S.M.; Protasio, J.; Riiff, T.; Hellerstein, M.K. Effect of bile acid sequestrants on glucose metabolism, hepatic de novo lipogenesis, and cholesterol and bile acid kinetics in type 2 diabetes: A randomised controlled study. Diabetologia 2012, 55, 432–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepherd, J. Mechanism of action of bile acid sequestrants and other lipid-lowering drugs. Cardiology 1989, 76, 65–74. [Google Scholar] [CrossRef]
- Björkhem, I.; Angelin, B.; Backman, L.; Liljeqvist, L.; Nilsell, K.; Einarsson, K. Triglyceride metabolism in human liver: Studies on hepatic phosphatidic-acid phosphatase in obese and non-obese subjects. Eur. J. Clin. Investig. 1984, 14, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Duell, P.B.; Gotto, A.M.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Flaim, J.A.; Ye, Z.; Catapano, A.L. Association of Bempedoic Acid Administration with Atherogenic Lipid Levels in Phase 3 Randomized Clinical Trials of Patients with Hypercholesterolemia. JAMA Cardiol. 2020, 5, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H.; Toth, P.; Weiss, S.; Mckenney, J.; Hunninghake, D.; Isaacsohn, J.; Donovan, J.M.; Burke, S.K. Low-dose combination therapy with colesevelam hydrochloride and lovastatin effectively decreases low-density lipoprotein cholesterol in patients with primary hypercholesterolemia. Clin. Cardiol. 2001, 24, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Knapp, H.H.; Schrott, H.; Ma, P.; Knopp, R.; Chin, B.; Gaziano, J.M.; Donovan, J.M.; Burke, S.K.; Davidson, M.H. Efficacy and safety of combination simvastatin and colesevelam in patients with primary hypercholesterolemia. Am. J. Med. 2001, 110, 352–360. [Google Scholar] [CrossRef]
- Alder, M.; Bavishi, A.; Zumpf, K.; Peterson, J.; Stone, N.J. A Meta-Analysis Assessing Additional LDL-C Reduction from Addition of a Bile Acid Sequestrant to Statin Therapy. Am. J. Med. 2020, 133, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Rigby, S.P.; Handelsman, Y.; Lai, Y.L.; Abby, S.L.; Tao, B.; Jones, M.R. Effects of colesevelam, rosiglitazone, or sitagliptin on glycemic control and lipid profile in patients with type 2 diabetes mellitus inadequately controlled by metformin monotherapy. Endocr. Pract. 2010, 16, 53–63. [Google Scholar] [CrossRef]
- Rosenstock, J.; Truitt, K.E.; Baz-Hecht, M.; Ford, D.M.; Tao, B.; Chou, H.S. Efficacy and safety of colesevelam in combination with pioglitazone in patients with type 2 diabetes mellitus. Horm. Metab. Res. 2014, 46, 943–949. [Google Scholar] [CrossRef]
- Bays, H. Colesevelam hydrochloride added to background metformin therapy in patients with type 2 diabetes mellitus: A pooled analysis from 3 clinical studies. Endocr. Pract. 2011, 17, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.B.; Rosenson, R.S.; Hernandez-Triana, E.; Misir, S.; Jones, M.R. Initial combination therapy with metformin plus colesevelam improves lipoprotein particles in patients with early type 2 diabetes mellitus. J. Clin. Lipidol. 2012, 6, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Sonne, D.P.; Mikkelsen, K.H.; Gluud, L.L.; Vilsbøll, T.; Knop, F.K. Bile acid sequestrants for glycemic control in patients with type 2 diabetes: A systematic review with meta-analysis of randomized controlled trials. J. Diabetes Complicat. 2017, 31, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirotri, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef] [Green Version]
- Rivers, S.M.; Kane, M.P.; Busch, R.S.; Bakst, G.; Hamilton, R.A. Colesevelam hydrochloride-ezetimibe combination lipid-lowering therapy in patients with diabetes or metabolic syndrome and a history of statin intolerance. Endocr. Pract. 2007, 13, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Annuzzi, G.; Corte, G.D.; Patti, L.; Cipriano, P.; Mangione, A.; Riccardi, G.; Rivellese, A.A. Ezetimibe beneficially influences fasting and postprandial triglyceride-rich lipoproteins in type 2 diabetes. Atherosclerosis 2011, 217, 142–148. [Google Scholar] [CrossRef]
- Nakou, E.S.; Filippatos, T.D.; Agouridis, A.P.; Kostara, C.; Bairaktari, E.T.; Elisaf, M.S. The Effects of ezetimibe and/or orlistat on triglyceride-rich lipoprotein metabolism in obese hypercholesterolemic patients. Lipids 2010, 45, 445–450. [Google Scholar] [CrossRef]
- Huijgen, R.; Abbink, E.J.; Bruckert, E.; Stalenhoef, A.F.H.; Imholz, B.P.M.; Durrington, P.N.; Trip, M.D.; Eriksson, M.; Visseren, F.L.J.; Schaefer, J.R.; et al. Colesevelam added to combination therapy with a statin and ezetimibe in patients with familial hypercholesterolemia: A 12-week, multicenter, randomized, double-blind, controlled trial. Clin. Ther. 2010, 32, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Miller, E.; Chitra, R. Efficacy and safety of rosuvastatin alone and in combination with cholestyramine in patients with severe hypercholesterolemia: A randomized, open-label, multicenter trial. Clin. Ther. 2004, 26, 1855–1864. [Google Scholar] [CrossRef]
- Karlson, B.W.; Palmer, M.K.; Nicholls, S.J.; Lundman, P.; Barter, P.J. A VOYAGER Meta-Analysis of the Impact of Statin Therapy on Low-Density Lipoprotein Cholesterol and Triglyceride Levels in Patients with Hypertriglyceridemia. Am. J. Cardiol. 2016, 117, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Nakou, E.S.; Filippatos, T.D.; Georgoula, M.; Kiortsis, D.N.; Tselepis, A.D.; Mikhailidis, D.P.; Elisaf, M.S. The effect of orlistat and ezetimibe, alone or in combination, on serum LDL and small dense LDL cholesterol levels in overweight and obese patients with hypercholesterolaemia. Curr. Med. Res. Opin. 2008, 24, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Al-Dury, S.; Marschall, H.U. Ileal bile acid transporter inhibition for the treatment of chronic constipation, cholestatic pruritus, and NASH. Front. Pharmacol. 2018, 9, 931. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, Q.; Li, Y.; Cao, S.; Zhang, Y.; Wang, Z.; Liu, G.; Li, J.; Gu, B. Apical sodium-dependent bile acid transporter, drug target for bile acid related diseases and delivery target for prodrugs: Current and future challenges. Pharmacol. Ther. 2020, 212, 107539. [Google Scholar] [CrossRef] [PubMed]
- Karpen, S.J.; Kelly, D.; Mack, C.; Stein, P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol. Int. 2020, 14, 677–689. [Google Scholar] [CrossRef]
- Simrén, M.; Bajor, A.; Gillberg, P.G.; Rudling, M.; Abrahamsson, H. Randomised clinical trial: The ileal bile acid transporter inhibitor A3309 vs. placebo in patients with chronic idiopathic constipation—A double-blind study. Aliment. Pharmacol. Ther. 2011, 34, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.S.; Camilleri, M.; McKinzie, S.; Burton, D.; Graffner, H.; Zinsmeister, A.R. Effects of A3309, an ileal bile acid transporter inhibitor, on colonic transit and symptoms in females with functional constipation. Am. J. Gastroenterol. 2011, 106, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Seki, M.; Taniguchi, S.; Ohta, A.; Gillberg, P.G.; Mattsson, J.P.; Camilleri, M. Safety and efficacy of elobixibat for chronic constipation: Results from a randomised, double-blind, placebo-controlled, phase 3 trial and an open-label, single-arm, phase 3 trial. Lancet Gastroenterol. Hepatol. 2018, 3, 537–547. [Google Scholar] [CrossRef]
- Kumagai, Y.; Amano, H.; Sasaki, Y.; Nakagawa, C.; Maeda, M.; Oikawa, I.; Furuie, H. Effect of single and multiple doses of elobixibat, an ileal bile acid transporter inhibitor, on chronic constipation: A randomized controlled trial. Br. J. Clin. Pharmacol. 2018, 84, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Nunez, D.J.; Yao, X.; Lin, J.; Walker, A.; Zuo, P.; Webster, L.; Krug-Gourley, S.; Zamek-Gliszczynski, M.J.; Gillmor, D.S.; Johnson, S.L. Glucose and lipid effects of the ileal apical sodium-dependent bile acid transporter inhibitor GSK2330672: Double-blind randomized trials with type 2 diabetes subjects taking metformin. Diabetes, Obes. Metab. 2016, 18, 654–662. [Google Scholar] [CrossRef]
- Graffner, H.; Gillberg, P.G.; Rikner, L.; Marschall, H.U. The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment. Pharmacol. Ther. 2016, 43, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Al-Dury, S.; Wahlström, A.; Wahlin, S.; Langedijk, J.; Elferink, R.O.; Ståhlman, M.; Marschall, H.U. Pilot study with IBAT inhibitor A4250 for the treatment of cholestatic pruritus in primary biliary cholangitis. Sci. Rep. 2018, 8, 6658. [Google Scholar] [CrossRef]
- Salmoirago-Blotcher, E.; Crawford, S.; Jackson, E.; Ockene, J.; Ockene, I. Constipation and risk of cardiovascular disease among postmenopausal women. Am. J. Med. 2011, 124, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenna, J.G.; Taskar, K.S.; Battista, C.; Bourdet, D.L.; Brouwer, K.L.R.; Brouwer, K.R.; Dai, D.; Funk, C.; Hafey, M.J.; Lai, Y.; et al. Can Bile Salt Export Pump Inhibition Testing in Drug Discovery and Development Reduce Liver Injury Risk? An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 916–932. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, K.; Hayashi, H.; Sabu, Y.; Tanikawa, K.; Fujishiro, J.; Kajikawa, D.; Wada, H.; Kudo, T.; Kage, M.; Kusuhara, H.; et al. Clinical phenotype and molecular analysis of a homozygous ABCB11 mutation responsible for progressive infantile cholestasis. J. Hum. Genet. 2018, 63, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Telbisz, Á.; Homolya, L. Recent advances in the exploration of the bile salt export pump (BSEP/ABCB11) function. Expert Opin. Ther. Targets 2016, 20, 501–514. [Google Scholar] [CrossRef] [Green Version]
- Gooijert, K.E.R.; Havinga, R.; Wolters, H.; Wang, R.; Ling, V.; Tazuma, S.; Verkade, H.J. The mechanism of increased biliary lipid secretion in mice with genetic inactivation of bile salt export pump. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G450–G457. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.S.; Kavesh, M.H.; Kriss, M.S.; Dewey, A.M.; Rinella, M.E.; Green, R.M. Hepatic overexpression of abcb11 promotes hypercholesterolemia and obesity in mice. Gastroenterology 2011, 141, 1404–1411.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, P.A.; Lan, T.; Rao, A. Bile acid transporters. J. Lipid Res. 2009, 50, 2340–2357. [Google Scholar] [CrossRef] [Green Version]
- Schaap, F.G.; van der Gaag, N.A.; Gouma, D.J.; Jansen, P.L.M. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology 2009, 49, 1228–1235. [Google Scholar] [CrossRef]
- Boyer, J.L.; Trauner, M.; Mennone, A.; Soroka, C.J.; Cai, S.Y.; Moustafa, T.; Zollner, G.; Lee, J.Y.; Ballatori, N. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTα-OSTβ in cholestasis in humans and rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1124–G1130. [Google Scholar] [CrossRef] [PubMed]
- Malinen, M.M.; Ali, I.; Bezençon, J.; Beaudoin, J.J.; Brouwer, K.L.R. Organic solute transporter OSTα/β is overexpressed in nonalcoholic steatohepatitis and modulated by drugs associated with liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G597–G609. [Google Scholar] [CrossRef] [PubMed]
- Ferslew, B.C.; Xie, G.; Johnston, C.K.; Su, M.; Stewart, P.W.; Jia, W.; Brouwer, K.L.R.; Sidney Barritt, A. Altered Bile Acid Metabolome in Patients with Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3318–3328. [Google Scholar] [CrossRef] [Green Version]
- Gao, E.; Cheema, H.; Waheed, N.; Mushtaq, I.; Erden, N.; Nelson-Williams, C.; Jain, D.; Soroka, C.J.; Boyer, J.L.; Khalil, Y.; et al. Organic Solute Transporter Alpha Deficiency: A Disorder With Cholestasis, Liver Fibrosis, and Congenital Diarrhea. Hepatology 2020, 71, 1879–1882. [Google Scholar] [CrossRef]
- Sultan, M.; Rao, A.; Elpeleg, O.; Vaz, F.M.; Abu-Libdeh, B.; Karpen, S.J.; Dawson, P.A. Organic solute transporter-β (SLC51B) deficiency in two brothers with congenital diarrhea and features of cholestasis. Hepatology 2018, 68, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Kim, K.H.; Lee, J.A.; Namkung, W.; Sun, A.Q.; Ananthanarayanan, M.; Suchy, F.J.; Shin, D.M.; Muallem, S.; Lee, M.G. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology 2002, 122, 1941–1953. [Google Scholar] [CrossRef]
- Slijepcevic, D.; Kaufman, C.; Wichers, C.G.K.; Gilglioni, E.H.; Lempp, F.A.; Duijst, S.; de Waart, D.R.; Elferink, R.P.J.O.; Mier, W.; Stieger, B.; et al. Impaired uptake of conjugated bile acids and hepatitis b virus pres1-binding in na(+) -taurocholate cotransporting polypeptide knockout mice. Hepatology 2015, 62, 207–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz, F.M.; Paulusma, C.C.; Huidekoper, H.; de Ru, M.; Lim, C.; Koster, J.; Ho-Mok, K.; Bootsma, A.H.; Groen, A.K.; Schaap, F.G.; et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: Conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015, 61, 260–267. [Google Scholar] [CrossRef]
- Deng, M.; Mao, M.; Guo, L.; Chen, F.P.; Wen, W.R.; Song, Y.Z. Clinical and molecular study of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Exp. Ther. Med. 2016, 12, 3294–3300. [Google Scholar] [CrossRef]
- Li, H.U.A.; Deng, M.E.I.; Guo, L.I.; Qiu, J.W.U.; Lin, G.U.I.Z.H.I.; Long, X.L.; Xiao, X.M.I.N.; Song, Y.Z. Clinical and molecular characterization of four patients with NTCP deficiency from two unrelated families harboring the novel SLC10A1 variant c.595A>C (p.Ser199Arg). Mol. Med. Rep. 2019, 20, 4915–4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkers, J.M.; Zehnder, B.; Van Westen, G.J.P.; Kwakkenbos, M.J.; Ijzerman, A.P.; Oude Elferink, R.P.J.; Beuers, U.; Urban, S.; Van De Graaf, S.F.J. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 2017, 7, 15307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkers, J.M.; Roscam Abbing, R.L.P.; van Weeghel, M.; Levels, J.H.M.; Boelen, A.; Schinkel, A.H.; Oude Elferink, R.P.J.; van de Graaf, S.F.J. Inhibition of Hepatic Bile Acid Uptake by Myrcludex B Promotes Glucagon-Like Peptide-1 Release and Reduces Obesity. CMGH 2020, 10, 451–466. [Google Scholar] [CrossRef]
- Blank, A.; Eidam, A.; Haag, M.; Hohmann, N.; Burhenne, J.; Schwab, M.; van de Graaf, S.F.J.; Meyer, M.R.; Maurer, H.H.; Meier, K.; et al. The NTCP-inhibitor Myrcludex B: Effects on Bile Acid Disposition and Tenofovir Pharmacokinetics. Clin. Pharmacol. Ther. 2018, 103, 341–348. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid x receptor agonist Obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Walters, J.R.F.; Johnston, I.M.; Nolan, J.D.; Vassie, C.; Pruzanski, M.E.; Shapiro, D.A. The response of patients with bile acid diarrhoea to the farnesoid x receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 2015, 41, 54–64. [Google Scholar] [CrossRef]
- Pencek, R.; Marmon, T.; Roth, J.D.; Liberman, A.; Hooshmand-Rad, R.; Young, M.A. Effects of obeticholic acid on lipoprotein metabolism in healthy volunteers. Diabetes Obes. Metab. 2016, 18, 936–940. [Google Scholar] [CrossRef]
- Pockros, P.J.; Fuchs, M.; Freilich, B.; Schiff, E.; Kohli, A.; Lawitz, E.J.; Hellstern, P.A.; Owens-Grillo, J.; Van Biene, C.; Shringarpure, R.; et al. CONTROL: A randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019, 39, 2082–2093. [Google Scholar] [CrossRef]
- Al-Khaifi, A.; Rudling, M.; Angelin, B. An FXR Agonist Reduces Bile Acid Synthesis Independently of Increases in FGF19 in Healthy Volunteers. Gastroenterology 2018, 155, 1012–1016. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.-S.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643. [Google Scholar] [CrossRef] [PubMed]
- Farr, S.; Stankovic, B.; Hoffman, S.; Masoudpoor, H.; Baker, C.; Taher, J.; Dean, A.E.; Anakk, S.; Adeli, K. Bile acid treatment and FXR agonism lower postprandial lipemia in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G682–G693. [Google Scholar] [CrossRef] [PubMed]
- Bass, J.Y.; Caldwell, R.D.; Caravella, J.A.; Chen, L.; Creech, K.L.; Deaton, D.N.; Madauss, K.P.; Marr, H.B.; McFadyen, R.B.; Miller, A.B.; et al. Substituted isoxazole analogs of farnesoid X receptor (FXR) agonist GW4064. Bioorg. Med. Chem. Lett. 2009, 19, 2969–2973. [Google Scholar] [CrossRef]
- Chiang, P.-C.; Thompson, D.C.; Ghosh, S.; Heitmeier, M.R. A formulation-enabled preclinical efficacy assessment of a farnesoid X receptor agonist, GW4064, in hamsters and cynomolgus monkeys. J. Pharm. Sci. 2011, 100, 4722–4733. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non-Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef]
- Wang, Y.; Crittenden, D.B.; Eng, C.; Zhang, Q.; Guo, P.; Chung, D.; Fenaux, M.; Klucher, K.; Jones, C.; Jin, F.; et al. Safety, Pharmacokinetics, Pharmacodynamics, and Formulation of Liver-Distributed Farnesoid X-Receptor Agonist TERN-101 in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 1198–1208. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Lee, K.-J.; Shim-Lopez, J.; Lee, J.; Wagner, B.; Smith, N.D.; Chen, H.C.; Lawitz, E.J. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 25–33. [Google Scholar] [CrossRef]
- Ratziu, V.; Rinella, M.E.; Neuschwander-Tetri, B.A.; Lawitz, E.; Denham, D.; Kayali, Z.; Sheikh, A.; Kowdley, K.V.; Desta, T.; Elkhashab, M.; et al. EDP-305 in patients with NASH: A phase II double-blind placebo-controlled dose-ranging study. J. Hepatol. 2021. [Google Scholar] [CrossRef]
- Kremoser, C. FXR agonists for NASH: How are they different and what difference do they make? J. Hepatol. 2021, 75, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Pols, T.W.H.; Nomura, M.; Harach, T.; Lo Sasso, G.; Oosterveer, M.H.; Thomas, C.; Rizzo, G.; Gioiello, A.; Adorini, L.; Pellicciari, R.; et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011, 14, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 2006, 191, 197–205. [Google Scholar] [CrossRef] [Green Version]
- Vassileva, G.; Golovko, A.; Markowitz, L.; Abbondanzo, S.J.; Zeng, M.; Yang, S.; Hoos, L.; Tetzloff, G.; Levitan, D.; Murgolo, N.J.; et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem. J. 2006, 398, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Donkers, J.M.; Roscam Abbing, R.L.P.; van de Graaf, S.F.J. Developments in bile salt based therapies: A critical overview. Biochem. Pharmacol. 2019, 161, 1–13. [Google Scholar] [CrossRef]
- Franke, A.; Balschun, T.; Karlsen, T.H.; Sventoraityte, J.; Nikolaus, S.; Mayr, G.; Domingues, F.S.; Albrecht, M.; Nothnagel, M.; Ellinghaus, D.; et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 2008, 40, 1319–1323. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Franke, A.; Melum, E.; Kaser, A.; Hov, J.R.; Balschun, T.; Lie, B.A.; Bergquist, A.; Schramm, C.; Weismüller, T.J.; et al. Genome-Wide Association Analysis in Primary Sclerosing Cholangitis. Gastroenterology 2010, 138, 1102–1111. [Google Scholar] [CrossRef]
- Hov, J.R.; Keitel, V.; Laerdahl, J.K.; Spomer, L.; Ellinghaus, E.; Elsharawy, A.; Melum, E.; Boberg, K.M.; Manke, T.; Balschun, T.; et al. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLoS ONE 2010, 5, e12403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, R.J.; Lin, J.; Vasist Johnson, L.S.; Gould, E.P.; Bowers, G.D.; Nunez, D.J. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 Agonist, SB-756050, in Type 2 Diabetes. Clin. Pharmacol. Drug Dev. 2013, 2, 213–222. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, L.; Anderson, J.; Ma, H.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Identification of novel pathways that control farnesoid X receptor-mediated hypocholesterolemia. J. Biol. Chem. 2010, 285, 3035–3043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozawa, E.; Amano, Y.; Yamakawa, H.; Haba, M.; Shimada, M.; Tozawa, R. Antidyslipidemic potential of a novel farnesoid X receptor antagonist in a hamster model of dyslipidemia: Comparative studies of other nonstatin agents. Pharmacol. Res. Perspect. 2018, 6, e00390. [Google Scholar] [CrossRef]
- Dong, B.; Young, M.; Liu, X.; Singh, A.B.; Liu, J. Regulation of lipid metabolism by obeticholic acid in hyperlipidemic hamsters. J. Lipid Res. 2017, 58, 350–363. [Google Scholar] [CrossRef] [Green Version]
- Bilz, S.; Samuel, V.; Morino, K.; Savage, D.; Choi, C.S.; Shulman, G.I. Activation of the farnesoid X receptor improves lipid metabolism in combined hyperlipidemic hamsters. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E716–E722. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, M.S.; Van Natta, M.L.; Connelly, M.A.; Vuppalanchi, R.; Neuschwander-Tetri, B.A.; Tonascia, J.; Guy, C.; Loomba, R.; Dasarathy, S.; Wattacheril, J.; et al. Impact of obeticholic acid on the lipoprotein profile in patients with non-alcoholic steatohepatitis. J. Hepatol. 2020, 72, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Borén, J.; Aguilar-Salinas, C.A.; Averna, M.; Ference, B.A.; Gaudet, D.; Hegele, R.A.; Kersten, S.; et al. Triglyceride-rich lipoproteins and their remnants: Metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur. Heart J. 2021, 42, 4791–4806. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| PMID | Therapy | Type of Patients | Number of Patient * | Year | LDL | Total Cholesterol | Non-HDL | HDL | Triglyceride | ApoB |
|---|---|---|---|---|---|---|---|---|---|---|
| 20047620 | Colestilan | T2D | 86 | 2010 |  |  |  | |||
| 27508319 | Cholestyramine | FH and CAD | 12 | 2016 |  |  |  |  |  | |
| 19789153 | Colesevelam | T2D | 56 | 2009 |  |  |  |  |  | |
| 19879596 | Colesevelam | Heterozygous FH | 63 | 2010 |  |  |  |  |  |  |
| 23152373 | Colesevelam | Prediabetes and primary hyperlipidaemia | 103 | 2012 |  |  |  |  |  |  |
| 24356792 | Colesevelam | T2D | 176 | 2013 |  |  |  |  |  | |
| BAS + Combination ** | ||||||||||
| 15639697 | Cholestyramine + Rosuvastatin | Severe hypercholesterolemia *** | 76 | 2004 |  |  |  |  |  | |
| 11403509 | Colesevelam + Lovastatin | Primary hypercholesterolemia | 27 | 2000 |  |  |  |  | ||
| 11286949 | Colesevelam + Simvastatin | Primary hypercholesterolemia | 34 | 2001 |  |  |  |  | ||
| 23170931 | Colesevelam + Rosuvastatin | Hypercholesterolemia and Impaired Fasting Glucose | 20 | 2013 |  |  |  |  |  | |
| 17360295 | Colesevelam + Ezetimibe | T2D with statin intolerance | 18 | 2007 |  |  |  |  |  | |
| 20435231 | Colesevelam + Ezetimibe + Statin | Familial Hypercholesterolemia | 44 | 2010 |  |  |  |  |  | |
| 21856592 | Colesevelam + Metformin | T2D | 355 | 2011 |  |  |  |  |  |  |
| 22836068 | Colesevelam + Metformin | T2D | 145 | 2012 |  |  |  |  |  | |
| 25054436 | Colesevelam + pioglitazone | T2D | 280 | 2014 |  |  |  |  |  |  |
| Decrease: |  |  |  |  | ||||||
| <10% | 10–20% | 20–30% | 30–40% | |||||||
| Increase: |  |  |  |  | ||||||
| <10% | 10–20% | >20% | No significance | |||||||
| Drugs | Mechanism of Action on Bile Acid Metabolism | Examples | Effects of Lipid Metabolism | Target Group | References |
|---|---|---|---|---|---|
| Bile acid sequestrants (BAS) | 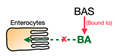 | Cholestyramine, Colesevelam, Colestilan | Total cholesterol ↓ LDL-C ↓ Triglyceride ↑ HDL-C variable * | Patients with dyslipidemia, T2D, and disturbed glucose metabolism | [61,62,63,64,65] |
| ASBT inhibitors | 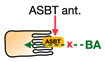 | Elobixibat, Linerixibat (GSK2330672), Odevixibat (A4250) ** | Total cholesterol ↓ LDL-C ↓ Triglyceride ↑ HDL-C = *** | Patients with chronic constipation, pruritus in primary biliary cholangitis, and T2D | [98,99,101,102] |
| FXR agonist (Steroidal) |  | Obeticholic acid | Total cholesterol ↑ LDL-C ↑ Triglyceride ↓/= HDL-C ↓ | Patients with NAFLD, NASH, T2D, and bile acid diarrhea | [126,127,128,129,130,156] |
| FXR agonists (Non-steroidal) | PX-102, Cilofexor, TERN-101, MET-409, GW4064 ** | LDL-C =/↑ **** Triglyceride = | Healthy volunteers Patients with NASH | [132,134,139,140] | |
| FXR agonist (Steroidal and non-steroidal) | EDP-305 | LDL-C ↑ Triglyceride = HDL-C ↓ | Patients with fibrotic NASH | [141] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Kuipers, F.; de Boer, J.F.; Kuivenhoven, J.A. Modulation of Bile Acid Metabolism to Improve Plasma Lipid and Lipoprotein Profiles. J. Clin. Med. 2022, 11, 4. https://doi.org/10.3390/jcm11010004
Zhang B, Kuipers F, de Boer JF, Kuivenhoven JA. Modulation of Bile Acid Metabolism to Improve Plasma Lipid and Lipoprotein Profiles. Journal of Clinical Medicine. 2022; 11(1):4. https://doi.org/10.3390/jcm11010004
Chicago/Turabian StyleZhang, Boyan, Folkert Kuipers, Jan Freark de Boer, and Jan Albert Kuivenhoven. 2022. "Modulation of Bile Acid Metabolism to Improve Plasma Lipid and Lipoprotein Profiles" Journal of Clinical Medicine 11, no. 1: 4. https://doi.org/10.3390/jcm11010004
APA StyleZhang, B., Kuipers, F., de Boer, J. F., & Kuivenhoven, J. A. (2022). Modulation of Bile Acid Metabolism to Improve Plasma Lipid and Lipoprotein Profiles. Journal of Clinical Medicine, 11(1), 4. https://doi.org/10.3390/jcm11010004