
Forgetting the Unforgettable: Transient Global Amnesia Part I: Pathophysiology and Etiology


Abstract
1. Introduction
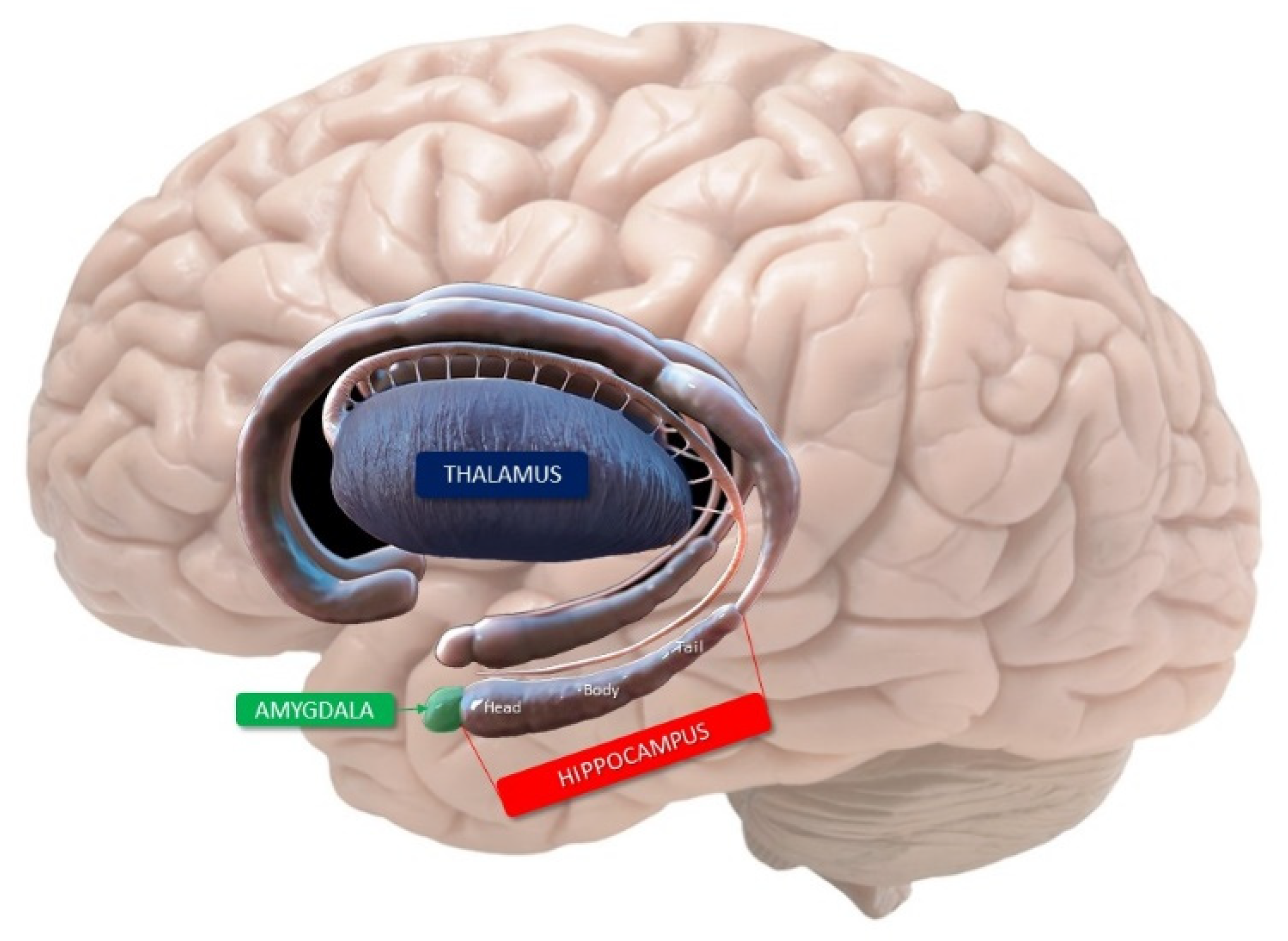
2. Anatomy of the Hippocampus
- Cornu ammonis (Hippocampus Proper)
- CA1 continues from the subiculum. Pyramidal somata are triangular, small and scattered.
- CA2 is composed of large, ovoid, densely packed somata, making the stratum piramidale dense and narrow, in sharp contrast to CA1.
- CA3 corresponds to the curve, or genu, of the cornu ammonis. Pyramidal somata are like those in CA2, but less numerous. A typical feature of this field is the presence of the mossy fibers, i.e., unmyelinated fibers arising from the gyrus dentatus.
- Dentate gyrus
3. Hippocampal Vascularization
- A.
- Main arterial supply of the hippocampus
- (a)
- PCA in its perimesencephalic P2 segment (situated in the crural and ambient cisterns) gives rise to the inferior temporal arteries (anterior, medial, and posterior) or Uchimura’s arteries, the posterolateral choroidal arteries, and the splenial arteries [21].
- (b)
- AchA, a branch of the internal carotid artery, on its way to the choroid plexuses of the temporal horn, gives rise to an uncal branch [22].
- (A)
- Mixed origin from AChA, PCA, and some branches of PCA such as the inferior temporal, lateral posterior choroidal, and splenial artery (57% of cases);
- (B)
- Origin from the inferior temporal branches of PCA (anterior, middle, posterior, and common inferior temporal trunk) (27% of cases);
- (C)
- Origin from the anterior inferior temporal branch of PCA (10% of cases);
- (D)
- Origin from the main trunk of the PCA (Uchimura artery) (3% of cases);
- (E)
- B.
- Superficial Hippocampal Arteries
- (a)
- Middle and posterior hippocampal arteries supply the hippocampal body and tail. Along the superficial hippocampal sulcus, the longitudinal terminal segments of these arteries form a dense network of anastomoses, from which originate the perforating arteries that enter the hippocampus between the indentations of the margo denticulatus. It has been hypothesized that the right-angle origin of the perforating arteries may explain the particular vulnerability of the hippocampus tissue to anoxia when there is a sudden drop in blood pressure [25].
- (b)
- Anterior hippocampal artery vascularizes the hippocampal head and uncus. The uncal branch of the AChA frequently anastomoses with the anterior hippocampal artery within the uncal sulcus, thus contributing to the vascularization of the hippocampal head [21].
- C.
- Intrahippocampal Arteries
- (a)
- Large Ventral Intrahippocampal Arteries vascularize CA1 and CA2.
- (b)
- Large Dorsal Intrahippocampal Arteries supply CA3 and sometimes CA2, as well as CA4 and the distal part of the dentate gyrus.
- (c)
- Small Ventral Intrahippocampal Arteries vascularize the proximal part of the gyrus dentatus.
- (d)
- Small Dorsal Intrahippocampal Arteries have a small intrahippocampal territory limited to CA3 and the adjacent part of CA4 [15].
- D.
- Venous circulation
4. Mnemonic Functions of Hippocampus
- (a)
- Episodic memory implies the ability to recall personal experiences and specific events framed in a personal temporal and spatial context;
- (b)
- The semantic memory includes all our knowledge of facts and concepts;
- (c)
- The polysynaptic pathway composes the following circuit: parietal, temporal, and occipital cortex → entorhinal cortex → dentate gyrus → CA3→ CA1→ subiculum → alveus → fimbria → fornix → mammillothalamic tract → anterior thalamus → posterior cingulated → retrosplenial cortex.
- The direct intra-hippocampal pathway is activated by afferent input from the temporal association cortex through the perirhinal and entorhinal area to CA1; from there, efferent projections reach the inferior temporal cortex, temporal pole, and prefrontal cortex through the subiculum and entorhinal cortex.
5. Etiology of TGA
5.1. Vascular Mechanism
- (a)
- Transient ischemic-hypoxic mechanism
- Clinically, the absence of associated focal neurologic dysfunction, such as lateralizing weakness and visual field deficits, during the TGA episode, is inconsistent with the ischemic hypothesis [4]. Furthermore, TIAs in the vast majority of cases last <60 min, with the bulk of these lasting only a few minutes [4,36,37]. TGA episodes, on the other hand, last on average 4–8 h, although a duration <1 h is not uncommon, ranging between 9–32% of cases [1,3,37,38].
- MRI studies showing in patients with TGA DWI changes in the hippocampal CA1 neuronal field, a region involved in the process of memory consolidation, provide support for the arterial ischemia hypothesis [3,4]. However, these changes are inconsistently present, reversible with time, and do not respect a clear arterial territory [3] (see Part II of this review for details). Furthermore, lesions associated with TGA are generally seen 24–72 h after symptom onset and disappear soon after; instead, symptoms of a clear ischemic nature are commonly associated with permanent lesions on MRI [3,4,44,45]. Finally, no abnormalities have been found in intracranial magnetic resonance angiography and in perfusion-weighted imaging during acute episodes, thus making the hypothesis of an arterial ischemia less likely [13,14,46,47].
- (b)
- Venous vascular mechanism
5.2. Migraine Related Mechanism
5.3. Epileptic Mechanism
5.4. Psychogenic Causes
6. Vulnerability to Hypoxia and Pathophysiology of Memory Disorders during TGA
- (a)
- (b)
- (c)
- The intrahippocampal territories of the deep blood arterial vessels show frequent variations, therefore, in most cases, CA1 is only vascularized by the large ventral intrahippocampal arteries, whereas CA2 and CA4 fields and the gyrus dentatus are vascularized by different arterial groups [74];
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hodges, J.R.; Warlow, C.P. Syndromes of transient amnesia: Towards a classification. A study of 153 cases. J. Neurol. Neurosurg. Psychiatry 1990, 53, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.R.; Warlow, C.P. The aetiology of transient global amnesia. A case-control study of 114 cases with prospective follow-up. Brain 1990, 113, 639–657. [Google Scholar] [CrossRef] [PubMed]
- Arena, J.E.; Rabinstein, A.A. Transient global amnesia. Mayo Clin. Proc. 2015, 90, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, D.R.; Smith, J.; Wade, R.R.; Cherukuru, N.; Ursani, A.; Dobruskina, Y.; Crist, T.; Busch, R.F.; Dhanani, R.M.; Dreyer, N. Transient global amnesia: Current perspectives. Neuropsychiatr. Dis. Treat. 2017, 13, 2691–2703. [Google Scholar] [CrossRef]
- Benon, R. Les ictus amnésiques dans les démences ‘organiques’. Ann. Méd. Psychol. 1909, 67, 207–219. [Google Scholar]
- Pearce, J.M.; Bogousslavsky, J. ‘Les ictus amnésiques’ and transient global amnesia. Eur. Neurol. 2009, 62, 188–192. [Google Scholar] [CrossRef]
- Hauge, T. Catheter vertebral angiography. Acta. Radiol. Suppl. 1954, 109, 1–219. [Google Scholar]
- Haas, D.C. Transient global amnesia after cerebral angiography. Arch. Neurol. 1983, 40, 258–259. [Google Scholar] [CrossRef]
- Bender, M.B. Syndrome of isolated episode of confusion with amnesia. J. Hillside Hosp. 1956, 5, 212–215. [Google Scholar]
- Guyotat, M.; Courjon, J. Les ictus amnésiques. J. Med. Lyon. 1956, 37, 697–701. [Google Scholar]
- Fisher, C.M.; Adams, R.D. Transient global amnesia. Trans. Am. Neurol. Assoc. 1958, 83, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.M.; Adams, R.D. Transient global amnesia. Acta Neurol. Scand. 1964, 40 (Suppl. S9), 1–83. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T.; Alfke, K.; Stingele, R.; Rohr, A.; Freitag-Wolf, S.; Jansen, O.; Deuschl, G. Selective affection of hippocampal CA-1 neurons in patients with transient global amnesia without long-term sequelae. Brain 2006, 129, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T.; Deuschl, G. Transient global amnesia: Functional anatomy and clinical implications. Lancet Neurol. 2010, 9, 205–214. [Google Scholar] [CrossRef]
- Duvernoy, H.; Cattin, F.; Risold, P.Y. Anatomy. In The Human Hippocampus, Functional Anatomy, Vascularization and Serial Sections with MRI, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 39–68. [Google Scholar] [CrossRef]
- Lorente de No, R. Studies on the structure of the cerebral cortex. II. Continuation of the study of the Ammonic system. J. Psychol. Neurol. 1934, 46, 113–177. [Google Scholar]
- Braak, H. On the structure of the human archicortex. I. The cornu ammonis. A Golgi and pigment architectonic study. Cell Tissue Res. 1974, 152, 349–383. [Google Scholar] [CrossRef] [PubMed]
- Mouritzen Dam, A. The density of neurons in the human hippocampus. Neuropathol. Appl. Neurobiol. 1979, 5, 249–264. [Google Scholar] [CrossRef]
- Marinković, S.; Milisavljević, M.; Puskas, L. Microvascular anatomy of the hippocampal formation. Surg. Neurol. 1992, 37, 339–349. [Google Scholar] [CrossRef]
- Erdem, A.; Yaşargil, G.; Roth, P. Microsurgical anatomy of the hippocampal arteries. J. Neurosurg. 1993, 79, 256–265. [Google Scholar] [CrossRef]
- Haegelen, C.; Berton, E.; Darnault, P.; Morandi, X. A revised classification of the temporal branches of the posterior cerebral artery. Surg. Radiol. Anat. 2012, 34, 385–391. [Google Scholar] [CrossRef]
- Morandi, X.; Brassier, G.; Darnault, P.; Mercier, P.; Scarabin, J.M.; Duval, J.M. Microsurgical anatomy of the anterior choroidal artery. Surg. Radiol. Anat. 1996, 18, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Spallazzi, M.; Dobisch, L.; Becke, A.; Berron, D.; Stucht, D.; Oeltze-Jafra, S.; Caffarra, P.; Speck, O.; Düzel, E. Hippocampal vascularization patterns: A high-resolution 7 Tesla time-of-flight magnetic resonance angiography study. Neuroimage Clin. 2019, 21, 101609. [Google Scholar] [CrossRef] [PubMed]
- Isolan, G.R.; Stefani, M.A.; Schneider, F.L.; Claudino, H.A.; Yu, Y.H.; Choi, G.G.; Telles, J.P.M.; Rabelo, N.N.; Figueiredo, E.G. Hippocampal vascularization: Proposal for a new classification. Surg. Neurol. Int. 2020, 11, 378. [Google Scholar] [CrossRef] [PubMed]
- Scharrer, E. Vascularization and vulnerability of the cornu ammonis in the opossum. Arch. Neurol. Psychiatry 1940, 44, 483–506. [Google Scholar] [CrossRef]
- Wolf, B.S.; Huang, Y.P. The subependymal veins of the lateral ventricles. Am. J. Roentgenol. Radium. Ther. Nucl. Med. 1964, 91, 406–426. [Google Scholar]
- von Bechterew, W. Demonstration eines Gehirns mit Zerstörung der vorderen und inneren Theile der Himrinde beider Schlafenlappen. Neurologisch. Zentralbl. 1900, 19, 990–999. [Google Scholar]
- Scoville, W.B.; Milner, B. Loss of recent memory after bilateral hippocampal lesions. J. Neurol. Neurosurg. Psychiatry 1957, 20, 11–21. [Google Scholar] [CrossRef]
- Markowitsch, H.J. Which brain regions are critically involved in the retrieval of old episodic memory? Brain Res. Brain Res. Rev. 1995, 21, 117–127. [Google Scholar] [CrossRef]
- Bird, C.M.; Burgess, N. The hippocampus and memory: Insights from spatial processing. Nat. Rev. Neurosci. 2008, 9, 182–194. [Google Scholar] [CrossRef]
- Kesner, R.P.; Morris, A.M.; Weeden, C.S.S. Spatial, temporal, and associative behavioral functions associated with different subregions of the hippocampus. In Oxford Handbook of Comparative Cognition; Zentall, T.R., Wasserman, E.A., Eds.; Oxford University Press: Oxford, NC, USA, 2012; pp. 322–346. [Google Scholar]
- Voss, J.L.; Bridge, D.J.; Cohen, N.J.; Walker, J.A. A Closer Look at the Hippocampus and Memory. Trends Cogn. Sci. 2017, 21, 577–588. [Google Scholar] [CrossRef]
- Squire, L.; Zola-Morgan, S.; Alvarez, P. Functional distinctions within the medical temporal lobe memory system: What is the evidence? Behav. Brain Sci. 1994, 17, 495–496. [Google Scholar] [CrossRef]
- Anand, K.S.; Dhikav, V. Hippocampus in health and disease: An overview. Ann. Indian Acad. Neurol. 2012, 15, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Bernal, I. Learning and memory consolidation: Linking molecular and behavioral data. Neuroscience 2011, 10, 176:129. [Google Scholar] [CrossRef] [PubMed]
- Quinette, P.; Guillery-Girard, B.; Dayan, J.; de la Sayette, V.; Marquis, S.; Viader, F.; Desgranges, B.; Eustache, F. What does transient global amnesia really mean? Review of the literature and thorough study of 142 cases. Brain 2006, 129, 1640–1658. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Minematsu, K.; Yasaka, M.; Wada, K.; Yamaguchi, T. The duration of symptoms in transient ischemic attack. Neurology 1999, 52, 976–980. [Google Scholar] [CrossRef]
- Romoli, M.; Tuna, M.A.; Li, L.; Paciaroni, M.; Giannandrea, D.; Tordo Caprioli, F.; Lotti, A.; Eusebi, P.; Mosconi, M.G.; Pellizzaro Venti, M.; et al. Time trends, frequency, characteristics and prognosis of short-duration transient global amnesia. Eur. J. Neurol. 2020, 27, 887–893. [Google Scholar] [CrossRef]
- Pantoni, L.; Bertini, E.; Lamassa, M.; Pracucci, G.; Inzitari, D. Clinical features, risk factors, and prognosis in transient global amnesia: A follow-up study. Eur. J. Neurol. 2005, 12, 350–356. [Google Scholar] [CrossRef]
- Melo, T.P.; Ferro, J.M.; Ferro, H. Transient global amnesia. A case control study. Brain 1992, 115, 261–270. [Google Scholar] [CrossRef]
- Zorzon, M.; Antonutti, L.; Masè, G.; Biasutti, E.; Vitrani, B.; Cazzato, G. Transient global amnesia and transient ischemic attack. Natural history, vascular risk factors, and associated conditions. Stroke 1995, 26, 1536–1542. [Google Scholar] [CrossRef]
- Schmidtke, K.; Ehmsen, L. Transient global amnesia and migraine. A case control study. Eur. Neurol. 1998, 40, 9–14. [Google Scholar] [CrossRef]
- Enzinger, C.; Thimary, F.; Kapeller, P.; Ropele, S.; Schmidt, R.; Ebner, F.; Fazekas, F. Transient global amnesia: Diffusion-weighted imaging lesions and cerebrovascular disease. Stroke 2008, 39, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Imamura, T.; Sakamoto, S.; Ishii, K.; Kazui, H.; Mori, E. Transient global amnesia: Increased signal intensity in the right hippocampus on diffusion-weighted magnetic resonance imaging. Neuroradiology 2002, 44, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Felix, M.M.; Castro, L.H.; Maia, A.C., Jr.; da Rocha, A.J. Evidence of acute ischemic tissue change in transient global amnesia in magnetic resonance imaging: Case report and literature review. J. Neuroimaging 2005, 15, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Budson, A.E.; Schlaug, G.; Briemberg, H.R. Perfusion- and diffusion-weighted magnetic resonance imaging in transient global amnesia. Neurology 1999, 53, 239–240. [Google Scholar] [CrossRef]
- Toledo, M.; Pujadas, F.; Grivé, E.; Alvarez-Sabin, J.; Quintana, M.; Rovira, A. Lack of evidence for arterial ischemia in transient global amnesia. Stroke 2008, 39, 476–479. [Google Scholar] [CrossRef]
- Lewis, S.L. Aetiology of transient global amnesia. Lancet 1998, 352, 397–399. [Google Scholar] [CrossRef]
- Akkawi, N.M.; Agosti, C.; Rozzini, L.; Anzola, G.P.; Padovani, A. Transient global amnesia and disturbance of venous flow patterns. Lancet 2001, 357, 957. [Google Scholar] [CrossRef]
- Han, K.; Hu, H.H.; Chao, A.C.; Chang, F.C.; Chung, C.P.; Hsu, H.Y.; Sheng, W.Y.; Wu, J. Transient Global Amnesia Linked to Impairment of Brain Venous Drainage: An Ultrasound Investigation. Front. Neurol. 2019, 10, 67. [Google Scholar] [CrossRef]
- Cejas, C.; Cisneros, L.F.; Lagos, R.; Zuk, C.; Ameriso, S.F. Internal jugular vein valve incompetence is highly prevalent in transient global amnesia. Stroke 2010, 41, 67–71. [Google Scholar] [CrossRef]
- Baracchini, C.; Tonello, S.; Farina, F.; Viaro, F.; Atzori, M.; Ballotta, E.; Manara, R. Jugular veins in transient global amnesia: Innocent bystanders. Stroke 2012, 43, 2289–2292. [Google Scholar] [CrossRef]
- Kang, Y.; Kim, E.; Kim, J.H.; Choi, B.S.; Jung, C.; Bae, Y.J.; Lee, K.M.; Lee, D.H. Time of flight MR angiography assessment casts doubt on the association between transient global amnesia and intracranial jugular venous reflux. Eur. Radiol. 2015, 25, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Sherzai, A.Z.; Ani, C.; Shavlik, D.; Ghamsary, M.; Lazar, E.; Sherzai, D. Strong Association Between Migraine and Transient Global Amnesia: A National Inpatient Sample Analysis. J Neuropsychiatry Clin. Neurosci. 2019, 31, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Liampas, I.; Siouras, A.S.; Siokas, V.; Tsouris, Z.; Rikos, D.; Brotis, A.; Aloizou, A.M.; Dastamani, M.; Dardiotis, E. Migraine in transient global amnesia: A meta-analysis of observational studies. J. Neurol. 2022, 269, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Jørgensen, M.B. Leao’s spreading depression in the hippocampus explains transient global amnesia. A hypothesis. Acta Neurol. Scand. 1986, 73, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Liampas, I.; Raptopoulou, M.; Siokas, V.; Bakirtzis, C.; Tsouris, Z.; Aloizou, A.M.; Dastamani, M.; Brotis, A.; Bogdanos, D.; Dardiotis, E. Conventional cardiovascular risk factors in Transient Global Amnesia: Systematic review and proposition of a novel hypothesis. Front. Neuroendocrinol. 2021, 61, 100909. [Google Scholar] [CrossRef] [PubMed]
- Pantoni, L.; Lamassa, M.; Inzitari, D. Transient global amnesia: A review emphasizing pathogenic aspects. Acta Neurol. Scand. 2000, 102, 275–283. [Google Scholar] [CrossRef]
- Miller, J.W.; Yanagihara, T.; Petersen, R.C.; Klass, D.W. Transient global amnesia and epilepsy. Electroencephalographic dis-tinction. Arch. Neurol. 1987, 44, 629–633. [Google Scholar] [CrossRef]
- Cole, A.J.; Gloor, P.; Kaplan, R. Transient global amnesia: The electroencephalogram at onset. Ann. Neurol. 1987, 22, 771–772. [Google Scholar] [CrossRef]
- Jacome, D.E. EEG features in transient global amnesia. Clin. Electroencephalogr. 1989, 20, 183–192. [Google Scholar] [CrossRef]
- Inzitari, D.; Pantoni, L.; Lamassa, M.; Pallanti, S.; Pracucci, G.; Marini, P. Emotional arousal and phobia in transient global amnesia. Arch. Neurol. 1997, 54, 866–873. [Google Scholar] [CrossRef]
- Spiegel, D.R.; Mccroskey, A.L.; Deyerle, B.A. A Case of Transient Global Amnesia: A Review and How It May Shed Further Insight into the Neurobiology of Delusions. Innov Clin. Neurosci 2016, 13, 32–41. [Google Scholar] [PubMed]
- Liampas, I.; Raptopoulou, M.; Mpourlios, S.; Siokas, V.; Tsouris, Z.; Aloizou, A.M.; Dastamani, M.; Brotis, A.; Bogdanos, D.; Xiromerisiou, G.; et al. Factors associated with recurrent transient global amnesia: Systematic review and pathophysiological insights. Rev. Neurosci. 2021, 32, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, C.; Higashida, K.; Fabbian, F.; De Giorgi, A.; Sandikci, V.; Ebert, A.; Platten, M.; Okazaki, S.; Manfredini, R.; Szabo, K. Chronobiology of transient global amnesia. J. Neurol. 2022, 269, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Alessandro, L.; Ricciardi, M.; Chaves, H.; Allegri, R.F. Acute amnestic syndromes. J. Neurol. Sci. 2020, 413, 116781. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, E.K. Selective Neuronal Vulnerability in the Hippocampus: Relationship to Neurological Diseases and Mechanisms for Differential Sensitivity of Neurons to Stress. In The Clinical Neurobiology of the Hippocampus; Michaelis, E.K., Bartsch, T., Eds.; Oxford University Press: Oxford, UK, 2012. [Google Scholar] [CrossRef]
- Sommer, W. Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch. Psychiatr. 1880, 10, 631–675. [Google Scholar] [CrossRef]
- Spielmeyer, W. Die Pathogenese des epileptischen Krampfes. Z. Dtsch Ges. Neurol. Psychiatr. 1927, 109, 501–520. [Google Scholar] [CrossRef]
- Rutecki, P.A.; Grossman, R.G.; Armstrong, D.; Irish-Loewen, S. Electrophysiological connections between the hippocampus and entorhinal cortex in patients with complex partial seizures. J. Neurosurg. 1989, 70, 667–675. [Google Scholar] [CrossRef]
- Kotapka, M.J.; Graham, D.I.; Adams, J.H.; Gennarelli, T.A. Hippocampal pathology in fatal human head injury without high intracranial pressure. J. Neurotrauma 1994, 11, 317–324. [Google Scholar] [CrossRef]
- Zola-Morgan, S.; Squire, L.R.; Rempel, N.L.; Clower, R.P.; Amaral, D.G. Enduring memory impairment in monkeys after ischemic damage to the hippocampus. J. Neurosci. 1992, 12, 2582–2596. [Google Scholar] [CrossRef]
- Kartsounis, L.D.; Rudge, P.; Stevens, J.M. Bilateral lesions of CA1 and CA2 fields of the hippocampus are sufficient to cause a severe amnesic syndrome in humans. J. Neurol. Neurosurg. Psychiatry 1995, 59, 95–98. [Google Scholar] [CrossRef]
- Uchimura, J. Über die Gefässversorgung des Ammons-hornes. Z. Gesamte Neurol. Psychiatr. 1928, 112, 1–19. [Google Scholar] [CrossRef]
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Charidimou, A.; Pantoni, L.; Love, S. The concept of sporadic cerebral small vessel disease: A road map on key definitions and current concepts. Int. J. Stroke 2016, 11, 6–18. [Google Scholar] [CrossRef]
- Cuadrado-Godia, E.; Dwivedi, P.; Sharma, S.; Ois Santiago, A.; Roquer Gonzalez, J.; Balcells, M.; Laird, J.; Turk, M.; Suri, H.S.; Nicolaides, A.; et al. Cerebral Small Vessel Disease: A Review Focusing on Pathophysiology, Biomarkers, and Machine Learning Strategies. J. Stroke 2018, 20, 302–320. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Krämer, L.M.; von Arnim, C.A.F.; Otto, M.; Thal, D.R. Capillary cerebral amyloid angiopathy in Alzheimer’s disease: Association with allocortical/hippocampal microinfarcts and cognitive decline. Acta Neuropathol. 2018, 135, 681–694. [Google Scholar] [CrossRef]
- Perosa, V.; Priester, A.; Ziegler, G.; Cardenas-Blanco, A.; Dobisch, L.; Spallazzi, M.; Assmann, A.; Maass, A.; Speck, O.; Oltmer, J.; et al. Hippocampal vascular reserve associated with cognitive performance and hippocampal volume. Brain 2020, 143, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Sapolsky, R.M. A mechanism for glucocorticoid toxicity in the hippocampus: Increased neuronal vulnerability to metabolic insults. J. Neurosci. 1985, 5, 1228–1232. [Google Scholar] [CrossRef]
- de Quervain, D.J.; Henke, K.; Aerni, A.; Treyer, V.; McGaugh, J.L.; Berthold, T.; Nitsch, R.M.; Buck, A.; Roozendaal, B.; Hock, C. Glucocorticoid-induced impairment of declarative memory retrieval is associated with reduced blood flow in the medial temporal lobe. Eur. J. Neurosci. 2003, 17, 1296–1302. [Google Scholar] [CrossRef]
- Dallman, M.F. Stress update Adaptation of the hypothalamic-pituitary-adrenal axis to chronic stress. Trends Endocrinol. Metab. 1993, 4, 62–69. [Google Scholar] [CrossRef]
- Gibbison, B.; Spiga, F.; Walker, J.J.; Russell, G.M.; Stevenson, K.; Kershaw, Y.; Zhao, Z.; Henley, D.; Angelini, G.D.; Lightman, S.L. Dynamic pituitary-adrenal interactions in response to cardiac surgery. Crit. Care Med. 2015, 43, 791–800. [Google Scholar] [CrossRef]
- Johren, O.; Inagami, T.; Saavedra, J.M. AT1A, AT1B, and AT2 angiotensin II receptor subtype gene expression in rat brain. Neuroreport 1995, 6, 2549–2552. [Google Scholar] [CrossRef]
- Wayner, M.J.; Polan-Curtain, J.; Armstrong, D.L. Dose and time dependency of angiotensin II inhibition of hippocampal long-term potentiation. Peptides 1995, 16, 1079–1082. [Google Scholar] [CrossRef]
- Armstrong, D.L.; Garcia, E.A.; Ma, T.; Quinones, B.; Wayner, M.J. Angiotensin II blockade of long-term potentiation at the perforant path–granule cell synapse in vitro. Peptides 1996, 17, 689–693. [Google Scholar] [CrossRef]
- Wright, J.W.; Reichert, J.R.; Davis, C.J.; Harding, J.W. Neural plasticity and the brain renin-angiotensin system. Neurosci. Biobehav. Rev. 2002, 26, 529–552. [Google Scholar] [CrossRef]
- Stillhard, G.; Landis, T.; Schiess, R.; Regard, M.; Sialer, G. Bitemporal hypoperfusion in transient global amnesia: 99m-Tc-HM-PAO SPECT and neuropsychological findings during and after an attack. J. Neurol. Neurosurg. Psychiatry 1990, 53, 339–342. [Google Scholar] [CrossRef]
- Goldenberg, G.; Podreka, I.; Pfaffelmeyer, N.; Wessely, P.; Deecke, L. Thalamic ischemia in transient global amnesia: A SPECT study. Neurology 1991, 41, 1748–1752. [Google Scholar] [CrossRef]
- Evans, J.; Wilson, B.; Wraight, E.P.; Hodges, J.R. Neuropsychological and SPECT scan findings during and after transient global amnesia: Evidence for the differential impairment of remote episodic memory. J. Neurol. Neurosurg. Psychiatry 1993, 56, 1227–1230. [Google Scholar] [CrossRef]
- Zeman, A.Z. Episodic memory in transient global amnesia. J. Neurol. Neurosurg. Psychiatry 1999, 66, 135. [Google Scholar] [CrossRef][Green Version]
- Jovin, T.G.; Vitti, R.A.; McCluskey, L.F. Evolution of temporal lobe hypoperfusion in transient global amnesia: A serial single photon emission computed tomography study. J. Neuroimaging 2000, 10, 238–241. [Google Scholar] [CrossRef]
- Park, Y.H.; Jang, J.W.; Yang, Y.; Kim, J.E.; Kim, S. Reflections of two parallel pathways between the hippocampus and neocortex in transient global amnesia: A cross-sectional study using DWI and SPECT. PLoS ONE 2013, 8, e67447. [Google Scholar] [CrossRef]
- Jang, J.W.; Park, Y.H.; Park, S.Y.; Wang, M.J.; Lim, J.S.; Kim, S.H.; Chun, I.K.; Yang, Y.; Kim, S. Longitudinal Cerebral Perfusion Change in Transient Global Amnesia Related to Left Posterior Medial Network Disruption. PLoS ONE 2015, 10, e0145658. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Cho, S.S.; Choi, J.Y.; Kim, Y.H. Transient global amnesia: A study with Tc-99m ECD SPECT shortly after symptom onset and after recovery. Diagn. Interv. Radiol. 2016, 22, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Baron, J.C.; Petit-Taboué, M.C.; Le Doze, F.; Desgranges, B.; Ravenel, N.; Marchal, G. Right frontal cortex hypometabolism in transient global amnesia. A PET study. Brain 1994, 117, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Eustache, F.; Desgranges, B.; Petit-Taboué, M.C.; de la Sayette, V.; Piot, V.; Sablé, C.; Marchal, G.; Baron, J.C. Transient global amnesia: Implicit/explicit memory dissociation and PET assessment of brain perfusion and oxygen metabolism in the acute stage. J. Neurol. Neurosurg. Psychiatry 1997, 63, 357–367. [Google Scholar] [CrossRef][Green Version]
- Peer, M.; Nitzan, M.; Goldberg, I.; Katz, J.; Gomori, J.M.; Ben-Hur, T.; Arzy, S. Reversible functional connectivity disturbances during transient global amnesia. Ann. Neurol. 2014, 75, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Zidda, F.; Griebe, M.; Ebert, A.; Ruttorf, M.; Roßmanith, C.; Gass, A.; Andoh, J.; Nees, F.; Szabo, K. Resting-state connectivity alterations during transient global amnesia. Neuroimage Clin. 2019, 23, 101869. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Lee, B.I.; Kim, S.E. Is Transient Global Amnesia a Network Disease? Eur. Neurol. 2018, 80, 345–354. [Google Scholar] [CrossRef]
- Hodel, J.; Leclerc, X.; Zuber, M.; Gerber, S.; Besson, P.; Marcaud, V.; Roubeau, V.; Brasme, H.; Ganzoui, I.; Ducreux, D.; et al. Structural Connectivity and Cortical Thickness Alterations in Transient Global Amnesia. AJNR Am. J. Neuroradiol. 2020, 41, 798–803. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Group | Origin of the Hippocampal Supply |
|---|---|
| A | AChA, the main trunk of the PCA, the inferior temporal, splenial, and lateral posterior choroidal branches of the PCA |
| B | All inferior temporal branches (anterior, middle, and posterior, and common inferior temporal trunk) of the PCA |
| C | Anterior inferior temporal branch of the PCA |
| D | PCA trunk (Uchimura artery) |
| E | AChA |
| F | Parieto-occipital artery, calcarine artery, and splenial artery |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sparaco, M.; Pascarella, R.; Muccio, C.F.; Zedde, M. Forgetting the Unforgettable: Transient Global Amnesia Part I: Pathophysiology and Etiology. J. Clin. Med. 2022, 11, 3373. https://doi.org/10.3390/jcm11123373
Sparaco M, Pascarella R, Muccio CF, Zedde M. Forgetting the Unforgettable: Transient Global Amnesia Part I: Pathophysiology and Etiology. Journal of Clinical Medicine. 2022; 11(12):3373. https://doi.org/10.3390/jcm11123373
Chicago/Turabian StyleSparaco, Marco, Rosario Pascarella, Carmine Franco Muccio, and Marialuisa Zedde. 2022. "Forgetting the Unforgettable: Transient Global Amnesia Part I: Pathophysiology and Etiology" Journal of Clinical Medicine 11, no. 12: 3373. https://doi.org/10.3390/jcm11123373
APA StyleSparaco, M., Pascarella, R., Muccio, C. F., & Zedde, M. (2022). Forgetting the Unforgettable: Transient Global Amnesia Part I: Pathophysiology and Etiology. Journal of Clinical Medicine, 11(12), 3373. https://doi.org/10.3390/jcm11123373