
Current and Innovated Managements for Autoimmune Bullous Skin Disorders: An Overview



Abstract
:1. Introduction
2. Therapeutic Managements
2.1. Intraepithelial Autoimmune Bullous Skin Disorders
2.1.1. Pemphigus Vulgaris (PV)
First-Line Therapies
Second-Line Therapies
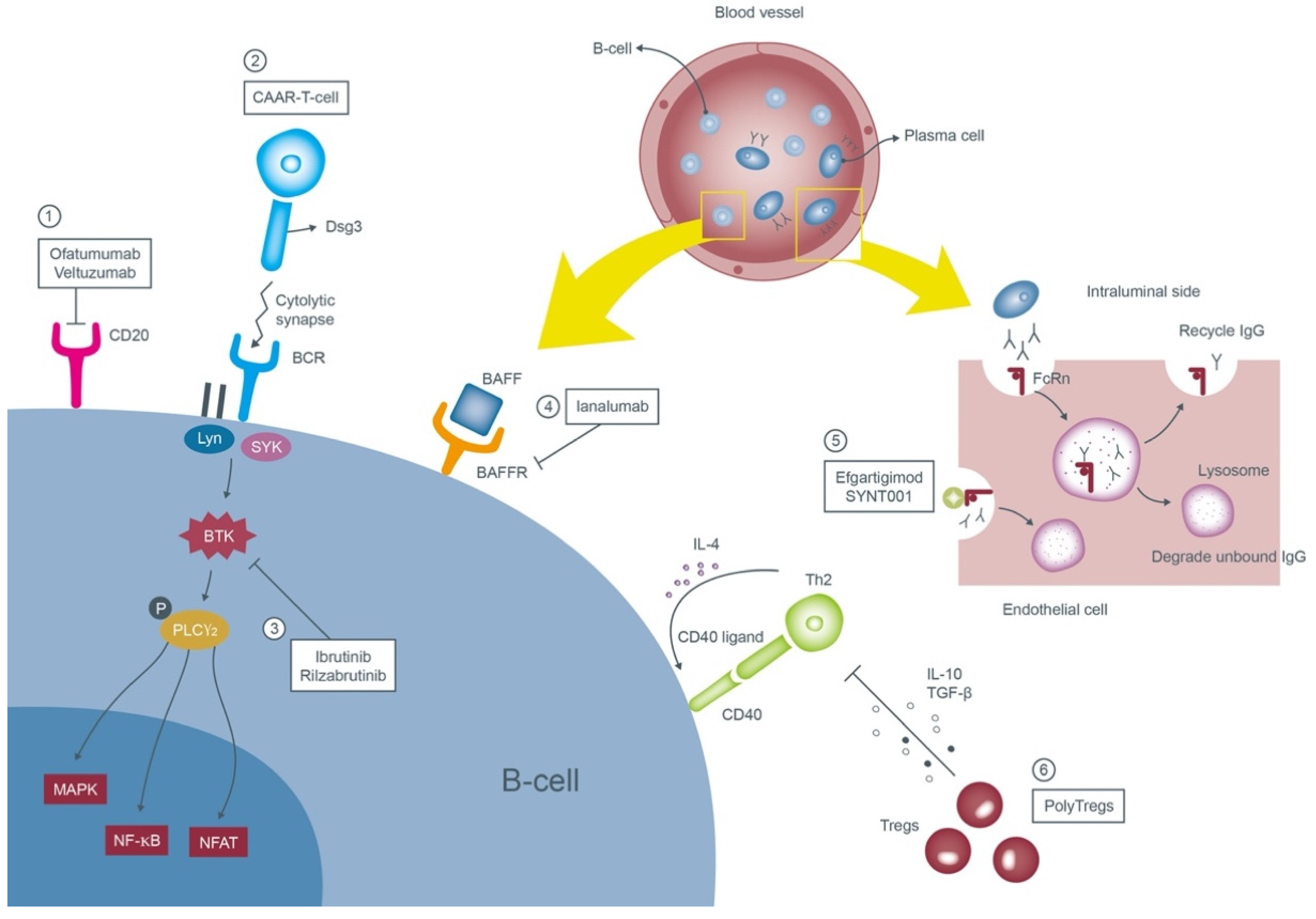
Emerging Options
2.1.2. Pemphigus Foliaceus (PF)
2.1.3. Pemphigus Erythematosus (PE)
2.1.4. IgA Pemphigus
2.1.5. Paraneoplastic Pemphigus (PNP)
Conventional Treatments
Emerging Options
2.2. Subepithelial Autoimmune Bullous Skin Disorders
2.2.1. Bullous Pemphigoid (BP)
First-Line Therapies
Second-Line Therapies
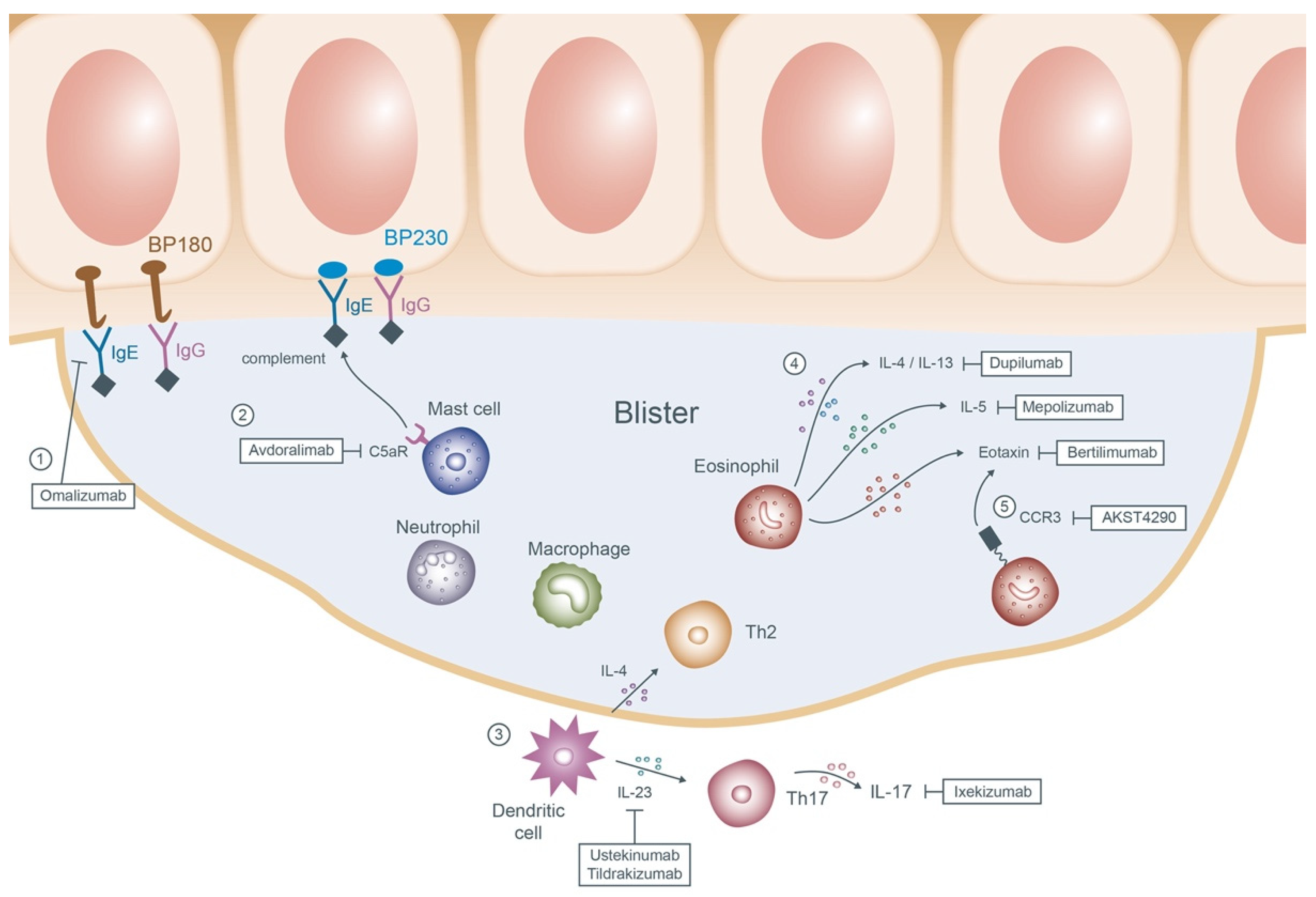
Emerging Options
2.2.2. Mucous Membrane Pemphigoid (MMP)
2.2.3. Linear IgA Bullous Dermatosis
2.2.4. Epidermolysis Bullosa Acquisita (EBA)
Conventional Treatments
Emerging Options
2.2.5. Dermatitis Herpetiformis (DH)
2.2.6. Laminin γ1 Pemphigoid
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kneisel, A.; Hertl, M. Autoimmune bullous skin diseases. Part 1: Clinical manifestations. J. Dtsch. Dermatol. Ges. 2011, 9, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Hertl, M.; Niedermeier, A.; Borradori, L. Autoimmune bullous skin disorders. Ther. Umschau. Rev. Ther. 2010, 67, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, N.; Zillikens, D.; Schmidt, E. Diagnosis of autoimmune bullous diseases. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2018, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saschenbrecker, S.; Karl, I.; Komorowski, L.; Probst, C.; Dähnrich, C.; Fechner, K.; Stöcker, W.; Schlumberger, W. Serological Diagnosis of Autoimmune Bullous Skin Diseases. Front. Immunol. 2019, 10, 1974. [Google Scholar] [CrossRef]
- Baum, S.; Sakka, N.; Artsi, O.; Trau, H.; Barzilai, A. Diagnosis and classification of autoimmune blistering diseases. Autoimmun. Rev. 2014, 13, 482–489. [Google Scholar] [CrossRef]
- Schmidt, E.; Kasperkiewicz, M.; Joly, P. Pemphigus. Lancet 2019, 394, 882–894. [Google Scholar] [CrossRef]
- Amerian, M.L.; Ahmed, A.R. Pemphigus erythematosus. Senear-Usher syndrome. Int. J. Dermatol. 1985, 24, 16–25. [Google Scholar] [CrossRef]
- Düker, I.; Schaller, J.; Rose, C.; Zillikens, D.; Hashimoto, T.; Kunze, J. Subcorneal pustular dermatosis-type IgA pemphigus with autoantibodies to desmocollins 1, 2, and 3. Arch. Dermatol. 2009, 145, 1159–1162. [Google Scholar] [CrossRef]
- Solimani, F.; Maglie, R.; Pollmann, R.; Schmidt, T.; Schmidt, A.; Ishii, N.; Tackenberg, B.; Kirschbaum, A.; Didona, D.; Pickert, J.; et al. Thymoma-Associated Paraneoplastic Autoimmune Multiorgan Syndrome—From Pemphigus to Lichenoid Dermatitis. Front. Immunol. 2019, 10, 1413. [Google Scholar] [CrossRef]
- Anhalt, G.J. Paraneoplastic pemphigus. J. Investig. Dermatol. Symp. Proc. 2004, 9, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, I.; Hodak, E.; Ackerman, L.; Mimouni, D.; Anhalt, G.J.; Calderon, S. Neoplasms associated with paraneoplastic pemphigus: A review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol. 2004, 40, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Ohzono, A.; Sogame, R.; Li, X.; Teye, K.; Tsuchisaka, A.; Numata, S.; Koga, H.; Kawakami, T.; Tsuruta, D.; Ishii, N.; et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br. J. Dermatol. 2015, 173, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.; Zillikens, D.; Schmidt, E. Diagnosis of Autoimmune Blistering Diseases. Front. Med. 2018, 5, 296. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.; della Torre, R.; Borradori, L. Clinical features and practical diagnosis of bullous pemphigoid. Dermatol. Clin. 2011, 29, 427–438, viii–ix. [Google Scholar] [CrossRef] [PubMed]
- Shetty, V.M.; Subramaniam, K.; Rao, R. Utility of immunofluorescence in dermatology. Indian Dermatol. Online J. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Chan, L.S.; Ahmed, A.R.; Anhalt, G.J.; Bernauer, W.; Cooper, K.D.; Elder, M.J.; Fine, J.D.; Foster, C.S.; Ghohestani, R.; Hashimoto, T.; et al. The first international consensus on mucous membrane pemphigoid: Definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch. Dermatol. 2002, 138, 370–379. [Google Scholar] [CrossRef]
- Thorne, J.E.; Anhalt, G.J.; Jabs, D.A. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology 2004, 111, 45–52. [Google Scholar] [CrossRef]
- Ahmed, A.R.; Kurgis, B.S.; Rogers, R.S., 3rd. Cicatricial pemphigoid. J. Am. Acad. Dermatol. 1991, 24, 987–1001. [Google Scholar] [CrossRef]
- Genovese, G.; Venegoni, L.; Fanoni, D.; Muratori, S.; Berti, E.; Marzano, A.V. Linear IgA bullous dermatosis in adults and children: A clinical and immunopathological study of 38 patients. Orphanet J. Rare Dis. 2019, 14, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roenigk, H.H., Jr.; Ryan, J.G.; Bergfeld, W.F. Epidermolysis bullosa acquisita. Report of three cases and review of all published cases. Arch. Dermatol. 1971, 103, 1–10. [Google Scholar] [CrossRef]
- Dainichi, T.; Kurono, S.; Ohyama, B.; Ishii, N.; Sanzen, N.; Hayashi, M.; Shimono, C.; Taniguchi, Y.; Koga, H.; Karashima, T.; et al. Anti-laminin gamma-1 pemphigoid. Proc. Natl. Acad. Sci USA 2009, 106, 2800–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kridin, K.; Ahmed, A.R. Anti-p200 Pemphigoid: A Systematic Review. Front. Immunol. 2019, 10, 2466. [Google Scholar] [CrossRef] [PubMed]
- Bishnoi, A.; De, D.; Handa, S.; Mahajan, R. Biologics in autoimmune bullous diseases: Current scenario. Indian J. Dermatol. Venereol. Leprol. 2021, 87, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Di Lernia, V.; Casanova, D.M.; Goldust, M.; Ricci, C. Pemphigus Vulgaris and Bullous Pemphigoid: Update on Diagnosis and Treatment. Dermatol. Pract. Concept. 2020, 10, e2020050. [Google Scholar] [CrossRef]
- Dumas, V.; Roujeau, J.C.; Wolkenstein, P.; Revuz, J.; Cosnes, A. The treatment of mild pemphigus vulgaris and pemphigus foliaceus with a topical corticosteroid. Br. J. Dermatol. 1999, 140, 1127–1129. [Google Scholar] [CrossRef]
- Bystryn, J.C.; Steinman, N.M. The adjuvant therapy of pemphigus: An update. Arch. Dermatol. 1996, 132, 203–212. [Google Scholar] [CrossRef]
- Santi, C.G.; Gripp, A.C.; Roselino, A.M.; Mello, D.S.; Gordilho, J.O.; Marsillac, P.F.; Porro, A.M. Consensus on the treatment of autoimmune bullous dermatoses: Bullous pemphigoid, mucous membrane pemphigoid and epidermolysis bullosa acquisita—Brazilian Society of Dermatology. An. Bras. Dermatol. 2019, 94, 33–47. [Google Scholar] [CrossRef]
- Eming, R.; Sticherling, M.; Hofmann, S.C.; Hunzelmann, N.; Kern, J.S.; Kramer, H.; Pfeiffer, C.; Schuster, V.; Zillikens, D.; Goebeler, M.; et al. S2k guidelines for the treatment of pemphigus vulgaris/foliaceus and bullous pemphigoid. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2015, 13, 833–844. [Google Scholar] [CrossRef]
- Rao, P.N.; Lakshmi, T.S. Pulse therapy and its modifications in pemphigus: A six year study. Indian J. Dermatol. Venereol. Leprol. 2003, 69, 329–333. [Google Scholar] [PubMed]
- Chams-Davatchi, C.; Esmaili, N.; Daneshpazhooh, M.; Valikhani, M.; Balighi, K.; Hallaji, Z.; Barzegari, M.; Akhyani, M.; Ghodsi, S.Z.; Seirafi, H.; et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J. Am. Acad. Dermatol. 2007, 57, 622–628. [Google Scholar] [CrossRef]
- Joly, P.; Maho-Vaillant, M.; Prost-Squarcioni, C.; Hebert, V.; Houivet, E.; Calbo, S.; Caillot, F.; Golinski, M.L.; Labeille, B.; Picard-Dahan, C.; et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): A prospective, multicentre, parallel-group, open-label randomised trial. Lancet 2017, 389, 2031–2040. [Google Scholar] [CrossRef]
- Huang, A.; Madan, R.K.; Levitt, J. Future therapies for pemphigus vulgaris: Rituximab and beyond. J. Am. Acad. Dermatol. 2016, 74, 746–753. [Google Scholar] [CrossRef]
- Cianchini, G.; Lupi, F.; Masini, C.; Corona, R.; Puddu, P.; De Pità, O. Therapy with rituximab for autoimmune pemphigus: Results from a single-center observational study on 42 cases with long-term follow-up. J. Am. Acad. Dermatol. 2012, 67, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Heelan, K.; Al-Mohammedi, F.; Smith, M.J.; Knowles, S.; Lansang, P.; Walsh, S.; Shear, N.H. Durable Remission of Pemphigus With a Fixed-Dose Rituximab Protocol. JAMA Dermatol. 2014, 150, 703–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, A.J.; Vinay, K.; Sawatkar, G.U.; Dogra, S.; Minz, R.W.; Shear, N.H.; Koga, H.; Ishii, N.; Hashimoto, T. Clinical and immunological outcomes of high- and low-dose rituximab treatments in patients with pemphigus: A randomized, comparative, observer-blinded study. Br. J. Dermatol. 2014, 170, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Smolen, J.S.; Betteridge, N.; Breedveld, F.C.; Burmester, G.; Dörner, T.; Ferraccioli, G.; Gottenberg, J.E.; Isaacs, J.; Kvien, T.K.; et al. Updated consensus statement on the use of rituximab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-J.; Wei, K.-C. Venous thromboembolism in a case with pemphigus vulgaris after infusion of rituximab plus systemic glucocorticoids and azathioprine: A possible adverse effect of rituximab? Derm. Sin. 2021, 39, 103–104. [Google Scholar] [CrossRef]
- Schiavo, A.L.; Puca, R.V.; Ruocco, V.; Ruocco, E. Adjuvant drugs in autoimmune bullous diseases, efficacy versus safety: Facts and controversies. Clin. Derm. 2010, 28, 337–343. [Google Scholar] [CrossRef]
- Hertl, M.; Jedlickova, H.; Karpati, S.; Marinovic, B.; Uzun, S.; Yayli, S.; Mimouni, D.; Borradori, L.; Feliciani, C.; Ioannides, D.; et al. Pemphigus. S2 Guideline for diagnosis and treatment—Guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J. Eur. Acad. Dermatol. Venereol. JEADV 2015, 29, 405–414. [Google Scholar] [CrossRef]
- Meurer, M. Immunosuppressive therapy for autoimmune bullous diseases. Clin. Derm. 2012, 30, 78–83. [Google Scholar] [CrossRef]
- Li, N.; Zhao, M.; Hilario-Vargas, J.; Prisayanh, P.; Warren, S.; Diaz, L.A.; Roopenian, D.C.; Liu, Z. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J. Clin. Investig. 2005, 115, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.H.O.; Enk, A.H. High-Dose Intravenous Immunoglobulin in Skin Autoimmune Disease. Front. Immunol. 2019, 10, 1090. [Google Scholar] [CrossRef] [PubMed]
- Daoud, Y.J.; Amin, K.G. Comparison of cost of immune globulin intravenous therapy to conventional immunosuppressive therapy in treating patients with autoimmune mucocutaneous blistering diseases. Int. Immunopharmacol. 2006, 6, 600–606. [Google Scholar] [CrossRef]
- Keskin, D.B.; Stern, J.N.; Fridkis-Hareli, M.; Razzaque Ahmed, A. Cytokine profiles in pemphigus vulgaris patients treated with intravenous immunoglobulins as compared to conventional immunosuppressive therapy. Cytokine 2008, 41, 315–321. [Google Scholar] [CrossRef]
- Lever, W.F.; Schaumburg-Lever, G. Treatment of pemphigus vulgaris. Results obtained in 84 patients between 1961 and 1982. Arch. Dermatol. 1984, 120, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Becker, B.A.; Gaspari, A.A. Pemphigus vulgaris and vegetans. Dermatol. Clin. 1993, 11, 429–452. [Google Scholar] [CrossRef]
- Ruocco, E.; Wolf, R.; Ruocco, V.; Brunetti, G.; Romano, F.; Lo Schiavo, A. Pemphigus: Associations and management guidelines: Facts and controversies. Clin. Derm. 2013, 31, 382–390. [Google Scholar] [CrossRef]
- Sinha, A.A.; Hoffman, M.B.; Janicke, E.C. Pemphigus vulgaris: Approach to treatment. Eur. J. Dermatol. EJD 2015, 25, 103–113. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Shimanovich, I.; Meier, M.; Schumacher, N.; Westermann, L.; Kramer, J.; Zillikens, D.; Schmidt, E. Treatment of severe pemphigus with a combination of immunoadsorption, rituximab, pulsed dexamethasone and azathioprine/mycophenolate mofetil: A pilot study of 23 patients. Br. J. Dermatol. 2012, 166, 154–160. [Google Scholar] [CrossRef]
- Zillikens, D.; Derfler, K.; Eming, R.; Fierlbeck, G.; Goebeler, M.; Hertl, M.; Hofmann, S.C.; Karlhofer, F.; Kautz, O.; Nitschke, M.; et al. Recommendations for the use of immunoapheresis in the treatment of autoimmune bullous diseases. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2007, 5, 881–887. [Google Scholar] [CrossRef]
- Behzad, M.; Möbs, C.; Kneisel, A.; Möller, M.; Hoyer, J.; Hertl, M.; Eming, R. Combined treatment with immunoadsorption and rituximab leads to fast and prolonged clinical remission in difficult-to-treat pemphigus vulgaris. Br. J. Dermatol. 2012, 166, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Robak, T.; Robak, E. New anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoid malignancies. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2011, 25, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Klufas, D.M.; Amerson, E.; Twu, O.; Clark, L.; Shinkai, K. Refractory pemphigus vulgaris successfully treated with ofatumumab. JAAD Case Rep. 2020, 6, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto-Immun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef]
- Ellebrecht, C.T.; Choi, E.J.; Allman, D.M.; Tsai, D.E.; Wegener, W.A.; Goldenberg, D.M.; Payne, A.S. Subcutaneous veltuzumab, a humanized anti-CD20 antibody, in the treatment of refractory pemphigus vulgaris. JAMA Derm. 2014, 150, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofford, L.J.; Nyhoff, L.E.; Sheehan, J.H.; Kendall, P.L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. [Google Scholar] [CrossRef] [Green Version]
- Corneth, O.B.J.; Klein Wolterink, R.G.J.; Hendriks, R.W. BTK Signaling in B Cell Differentiation and Autoimmunity. Curr. Top. Microbiol. Immunol. 2016, 393, 67–105. [Google Scholar] [CrossRef]
- Campbell, R.; Chong, G.; Hawkes, E.A. Novel Indications for Bruton’s Tyrosine Kinase Inhibitors, beyond Hematological Malignancies. J. Clin. Med. 2018, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Didona, D.; Maglie, R.; Eming, R.; Hertl, M. Pemphigus: Current and Future Therapeutic Strategies. Front. Immunol. 2019, 10, 1418. [Google Scholar] [CrossRef] [Green Version]
- Patsatsi, A.; Murrell, D.F. Bruton Tyrosine Kinase Inhibition and Its Role as an Emerging Treatment in Pemphigus. Front. Med. 2021, 8, 708071. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Sandhu, S.; Imlay-Gillespie, L.; Mulligan, S.; Shumack, S. Successful use of Bruton’s kinase inhibitor, ibrutinib, to control paraneoplastic pemphigus in a patient with paraneoplastic autoimmune multiorgan syndrome and chronic lymphocytic leukaemia. Australas J. Derm. 2017, 58, e240–e242. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Crespo, A.; Rodriguez, V.; Matas-Cespedes, A.; Lee, E.; Rivas-Delgado, A.; Giné, E.; Navarro, A.; Beà, S.; Campo, E.; López-Guillermo, A.; et al. The Bruton tyrosine kinase inhibitor CC-292 shows activity in mantle cell lymphoma and synergizes with lenalidomide and NIK inhibitors depending on nuclear factor-κB mutational status. Haematologica 2017, 102, e447–e451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, D.F.; Patsatsi, A.; Stavropoulos, P.; Baum, S.; Zeeli, T.; Kern, J.S.; Roussaki-Schulze, A.V.; Sinclair, R.; Bassukas, I.D.; Thomas, D.; et al. Proof of concept for the clinical effects of oral rilzabrutinib, the first Bruton tyrosine kinase inhibitor for pemphigus vulgaris: The phase II BELIEVE study. Br. J. Dermatol. 2021, 185, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Bieber, K.; Ludwig, R.J. Current Clinical Trials in Pemphigus and Pemphigoid. Front. Immunol. 2019, 10, 978. [Google Scholar] [CrossRef]
- Mackay, F.; Browning, J.L. BAFF: A fundamental survival factor for B cells. Nat. Rev. Immunol. 2002, 2, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Lesley, R.; Xu, Y.; Kalled, S.L.; Hess, D.M.; Schwab, S.R.; Shu, H.B.; Cyster, J.G. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 2004, 20, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Hasegawa, M.; Matsushita, Y.; Echigo, T.; Wayaku, T.; Horikawa, M.; Ogawa, F.; Takehara, K.; Sato, S. Elevated serum BAFF levels in patients with localized scleroderma in contrast to other organ-specific autoimmune diseases. Exp. Derm. 2007, 16, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Groom, J.; Kalled, S.L.; Cutler, A.H.; Olson, C.; Woodcock, S.A.; Schneider, P.; Tschopp, J.; Cachero, T.G.; Batten, M.; Wheway, J.; et al. Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren’s syndrome. J. Clin. Investig. 2002, 109, 59–68. [Google Scholar] [CrossRef]
- Cheema, G.S.; Roschke, V.; Hilbert, D.M.; Stohl, W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001, 44, 1313–1319. [Google Scholar] [CrossRef]
- Kuo, T.T.; Baker, K.; Yoshida, M.; Qiao, S.W.; Aveson, V.G.; Lencer, W.I.; Blumberg, R.S. Neonatal Fc receptor: From immunity to therapeutics. J. Clin. Immunol. 2010, 30, 777–789. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.F., Jr.; Bril, V.; Burns, T.M.; Mantegazza, R.; Bilinska, M.; Szczudlik, A.; Beydoun, S.; Garrido, F.; Piehl, F.; Rottoli, M.; et al. Randomized phase 2 study of FcRn antagonist efgartigimod in generalized myasthenia gravis. Neurology 2019, 92, e2661–e2673. [Google Scholar] [CrossRef]
- Newland, A.C.; Sánchez-González, B.; Rejtő, L.; Egyed, M.; Romanyuk, N.; Godar, M.; Verschueren, K.; Gandini, D.; Ulrichts, P.; Beauchamp, J.; et al. Phase 2 study of efgartigimod, a novel FcRn antagonist, in adult patients with primary immune thrombocytopenia. Am. J. Hematol. 2020, 95, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Willenborg, S.; Hünig, T.; Deeg, C.A.; Sonderstrup, G.; Hertl, M.; Eming, R. Induction of T regulatory cells by the superagonistic anti-CD28 antibody D665 leads to decreased pathogenic IgG autoantibodies against desmoglein 3 in a HLA-transgenic mouse model of pemphigus vulgaris. Exp. Derm. 2016, 25, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Zwang, N.A.; Leventhal, J.R. Cell Therapy in Kidney Transplantation: Focus on Regulatory T Cells. J. Am. Soc. Nephrol. 2017, 28, 1960–1972. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-E.; Wu, R.-W.; Chang, C.-H. An elderly female with pemphigus foliaceus possibly induced by losartan/hydrochlorothiazide. Derm. Sin. 2021, 39, 107–108. [Google Scholar] [CrossRef]
- Kasperkiewicz, M.; Ellebrecht, C.T.; Takahashi, H.; Yamagami, J.; Zillikens, D.; Payne, A.S.; Amagai, M. Pemphigus. Nat. Rev. Dis. Primers 2017, 3, 17026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, S.C.; Juratli, H.A.; Eming, R. Bullous autoimmune dermatoses. J. Dtsch. Dermatol. Ges. J. Ger. Soc. Dermatol. JDDG 2018, 16, 1339–1358. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Chang, T.; Wu, C.; Chang, Y. Intraepidermal neutrophilic dermatosis-type immunoglobulin A pemphigus. Derm. Sin. 2021, 39, 47–48. [Google Scholar] [CrossRef]
- Gruss, C.; Zillikens, D.; Hashimoto, T.; Amagai, M.; Kroiss, M.; Vogt, T.; Landthaler, M.; Stolz, W. Rapid response of IgA pemphigus of subcorneal pustular dermatosis type to treatment with isotretinoin. J. Am. Acad. Dermatol. 2000, 43, 923–926. [Google Scholar] [CrossRef]
- Ruiz-Genao, D.P.; Hernández-Núñez, A.; Hashimoto, T.; Amagai, M.; Fernández-Herrera, J.; García-Díez, A. A case of IgA pemphigus successfully treated with acitretin. Br. J. Dermatol. 2002, 147, 1040–1042. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.M.; Bessinger, G.T.; Altman, C.E.; Belnap, C.M. Rapid response of IgA pemphigus of the subcorneal pustular dermatosis subtype to treatment with adalimumab and mycophenolate mofetil. J. Am. Acad. Dermatol. 2005, 53, 541–543. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, V.N.; Srivastava, G. Paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome. Int. J. Dermatol. 2009, 48, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, M.; Itin, P.; Wernli, M.; Borradori, L.; Bargetzi, M.J. Successful treatment of paraneoplastic pemphigus in follicular NHL with rituximab: Report of a case and review of treatment for paraneoplastic pemphigus in NHL and CLL. Am. J. Hematol. 2001, 66, 142–144. [Google Scholar] [CrossRef]
- Zhao, Y.; Su, H.; Shen, X.; Du, J.; Zhang, X.; Zhao, Y. The immunological function of CD52 and its targeting in organ transplantation. Inflamm. Res. 2017, 66, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Ruck, T.; Bittner, S.; Wiendl, H.; Meuth, S.G. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 2015, 16, 16414–16439. [Google Scholar] [CrossRef]
- Paolino, G.; Didona, D.; Magliulo, G.; Iannella, G.; Didona, B.; Mercuri, S.R.; Moliterni, E.; Donati, M.; Ciofalo, A.; Granata, G.; et al. Paraneoplastic Pemphigus: Insight into the Autoimmune Pathogenesis, Clinical Features and Therapy. Int. J. Mol. Sci. 2017, 18, 2532. [Google Scholar] [CrossRef] [Green Version]
- Hohwy, T.; Bang, K.; Steiniche, T.; Peterslund, N.A.; d’Amore, F. Alemtuzumab-induced remission of both severe paraneoplastic pemphigus and leukaemic bone marrow infiltration in a case of treatment-resistant B-cell chronic lymphocytic leukaemia. Eur. J. Haematol. 2004, 73, 206–209. [Google Scholar] [CrossRef]
- Bech, R.; Baumgartner-Nielsen, J.; Peterslund, N.A.; Steiniche, T.; Deleuran, M.; d’Amore, F. Alemtuzumab is effective against severe chronic lymphocytic leukaemia-associated paraneoplastic pemphigus. Br. J. Dermatol. 2013, 169, 469–472. [Google Scholar] [CrossRef]
- Joly, P.; Roujeau, J.C.; Benichou, J.; Picard, C.; Dreno, B.; Delaporte, E.; Vaillant, L.; D’Incan, M.; Plantin, P.; Bedane, C.; et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N. Engl. J. Med. 2002, 346, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Joly, P.; Roujeau, J.C.; Benichou, J.; Delaporte, E.; D’Incan, M.; Dreno, B.; Bedane, C.; Sparsa, A.; Gorin, I.; Picard, C.; et al. A comparison of two regimens of topical corticosteroids in the treatment of patients with bullous pemphigoid: A multicenter randomized study. J. Invest. Derm. 2009, 129, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirtschig, G.; Middleton, P.; Bennett, C.; Murrell, D.F.; Wojnarowska, F.; Khumalo, N.P. Interventions for bullous pemphigoid. Cochrane Database Syst. Rev. 2010, 2010, Cd002292. [Google Scholar] [CrossRef] [PubMed]
- Daniel, B.S.; Borradori, L.; Hall, R.P., 3rd; Murrell, D.F. Evidence-based management of bullous pemphigoid. Dermatol. Clin. 2011, 29, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Kawai, K. Doxycycline as an initial treatment of bullous pemphigoid in Japanese patients. J. Cutan. Immunol. Allergy 2020, 3, 80–85. [Google Scholar] [CrossRef]
- Van Beek, N.; Lüttmann, N.; Huebner, F.; Recke, A.; Karl, I.; Schulze, F.S.; Zillikens, D.; Schmidt, E. Correlation of Serum Levels of IgE Autoantibodies Against BP180 With Bullous Pemphigoid Disease Activity. JAMA Dermatol. 2017, 153, 30–38. [Google Scholar] [CrossRef]
- Yu, K.K.; Crew, A.B.; Messingham, K.A.; Fairley, J.A.; Woodley, D.T. Omalizumab therapy for bullous pemphigoid. J. Am. Acad. Dermatol. 2014, 71, 468–474. [Google Scholar] [CrossRef]
- Balakirski, G.; Alkhateeb, A.; Merk, H.F.; Leverkus, M.; Megahed, M. Successful treatment of bullous pemphigoid with omalizumab as corticosteroid-sparing agent: Report of two cases and review of literature. J. Eur. Acad. Dermatol. Venereol. JEADV 2016, 30, 1778–1782. [Google Scholar] [CrossRef]
- De, D.; Kaushik, A.; Handa, S.; Mahajan, R.; Schmidt, E. Omalizumab: An underutilized treatment option in bullous pemphigoid patients with co-morbidities. J. Eur. Acad. Dermatol. Venereol. JEADV 2021, 35, e469–e472. [Google Scholar] [CrossRef]
- Heimbach, L.; Li, Z.; Berkowitz, P.; Zhao, M.; Li, N.; Rubenstein, D.S.; Diaz, L.A.; Liu, Z. The C5a receptor on mast cells is critical for the autoimmune skin-blistering disease bullous pemphigoid. J. Biol. Chem. 2011, 286, 15003–15009. [Google Scholar] [CrossRef] [Green Version]
- Plée, J.; Le Jan, S.; Giustiniani, J.; Barbe, C.; Joly, P.; Bedane, C.; Vabres, P.; Truchetet, F.; Aubin, F.; Antonicelli, F.; et al. Integrating longitudinal serum IL-17 and IL-23 follow-up, along with autoantibodies variation, contributes to predict bullous pemphigoid outcome. Sci. Rep. 2015, 5, 18001. [Google Scholar] [CrossRef] [PubMed]
- Wakugawa, M.; Nakamura, K.; Hino, H.; Toyama, K.; Hattori, N.; Okochi, H.; Yamada, H.; Hirai, K.; Tamaki, K.; Furue, M. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: Correlation with tissue eosinophilia. Br. J. Dermatol. 2000, 143, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Rico, M.J.; Benning, C.; Weingart, E.S.; Streilein, R.D.; Hall, R.P., 3rd. Characterization of skin cytokines in bullous pemphigoid and pemphigus vulgaris. Br. J. Dermatol. 1999, 140, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, P.; Caproni, M.; Berti, S.; Bianchi, B.; Amato, L.; De Pità, O.; Frezzolini, A. The role of T lymphocytes and cytokines in the pathogenesis of pemphigoid gestationis. Br. J. Dermatol. 2003, 148, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Abdat, R.; Waldman, R.A.; de Bedout, V.; Czernik, A.; McLeod, M.; King, B.; Gordon, S.; Ahmed, R.; Nichols, A.; Rothe, M.; et al. Dupilumab as a novel therapy for bullous pemphigoid: A multicenter case series. J. Am. Acad. Dermatol. 2020, 83, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Maglie, R.; Hertl, M. Pharmacological advances in pemphigoid. Curr. Opin. Pharm. 2019, 46, 34–43. [Google Scholar] [CrossRef]
- Simon, D.; Yousefi, S.; Cazzaniga, S.; Bürgler, C.; Radonjic, S.; Houriet, C.; Heidemeyer, K.; Klötgen, H.W.; Kozlowski, E.; Borradori, L.; et al. Mepolizumab failed to affect bullous pemphigoid: A randomized, placebo-controlled, double-blind phase 2 pilot study. Allergy 2020, 75, 669–672. [Google Scholar] [CrossRef]
- Grimaldi, J.C.; Yu, N.X.; Grunig, G.; Seymour, B.W.; Cottrez, F.; Robinson, D.S.; Hosken, N.; Ferlin, W.G.; Wu, X.; Soto, H.; et al. Depletion of eosinophils in mice through the use of antibodies specific for C-C chemokine receptor 3 (CCR3). J. Leukoc. Biol. 1999, 65, 846–853. [Google Scholar] [CrossRef]
- Taylor, J.; McMillan, R.; Shephard, M.; Setterfield, J.; Ahmed, R.; Carrozzo, M.; Grando, S.; Mignogna, M.; Kuten-Shorrer, M.; Musbah, T.; et al. World Workshop on Oral Medicine VI: A systematic review of the treatment of mucous membrane pemphigoid. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2015, 120, 161–171.e120. [Google Scholar] [CrossRef]
- Lee, H.Y.; Blazek, C.; Beltraminelli, H.; Borradori, L. Oral mucous membrane pemphigoid: Complete response to topical tacrolimus. Acta Derm. Venereol. 2011, 91, 604–605. [Google Scholar] [CrossRef] [Green Version]
- Ujiie, H.; Iwata, H.; Yamagami, J.; Nakama, T.; Aoyama, Y.; Ikeda, S.; Ishii, N.; Iwatsuki, K.; Kurosawa, M.; Sawamura, D.; et al. Japanese guidelines for the management of pemphigoid (including epidermolysis bullosa acquisita). J. Derm. 2019, 46, 1102–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alajlan, A.; Al-Khawajah, M.; Al-Sheikh, O.; Al-Saif, F.; Al-Rasheed, S.; Al-Hoqail, I.; Hamadah, I.R. Treatment of linear IgA bullous dermatosis of childhood with flucloxacillin. J. Am. Acad. Dermatol. 2006, 54, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Gürcan, H.M.; Ahmed, A.R. Current concepts in the treatment of epidermolysis bullosa acquisita. Expert Opin. Pharm. 2011, 12, 1259–1268. [Google Scholar] [CrossRef]
- Sami, N. Mycophenolate mofetil (MMF) in the treatment of epidermolysis bullosa acquisita (EBA) long-term follow-up. JAAD Case Rep. 2015, 1, 321–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, J.B.; Yancey, K.B.; Engler, R.J.; James, W.D. Epidermolysis bullosa acquisita: Efficacy of high-dose intravenous immunoglobulins. J. Am. Acad. Dermatol. 1994, 31, 827–828. [Google Scholar] [CrossRef]
- Sadler, E.; Schafleitner, B.; Lanschuetzer, C.; Laimer, M.; Pohla-Gubo, G.; Hametner, R.; Hintner, H.; Bauer, J.W. Treatment-resistant classical epidermolysis bullosa acquisita responding to rituximab. Br. J. Dermatol. 2007, 157, 417–419. [Google Scholar] [CrossRef]
- Egan, C.A.; Brown, M.; White, J.D.; Yancey, K.B. Treatment of epidermolysis bullosa acquisita with the humanized anti-Tac mAb daclizumab. Clin. Immunol. 2001, 101, 146–151. [Google Scholar] [CrossRef]
- Fry, L. Dermatitis herpetiformis: Problems, progress and prospects. Eur. J. Dermatol. EJD 2002, 12, 523–531. [Google Scholar]
- McFadden, J.P.; Leonard, J.N.; Powles, A.V.; Rutman, A.J.; Fry, L. Sulphamethoxypyridazine for dermatitis herpetiformis, linear IgA disease and cicatricial pemphigoid. Br. J. Dermatol. 1989, 121, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Willsteed, E.; Lee, M.; Wong, L.C.; Cooper, A. Sulfasalazine and dermatitis herpetiformis. Australas J. Derm. 2005, 46, 101–103. [Google Scholar] [CrossRef]
- Wozniak, K.; Hashimoto, T.; Fukuda, S.; Ohyama, B.; Ishii, N.; Koga, H.; Dainichi, T.; Kowalewski, C. IgA Anti-p200 Pemphigoid. Arch. Dermatol. 2011, 147, 1306–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goletz, S.; Hashimoto, T.; Zillikens, D.; Schmidt, E. Anti-p200 pemphigoid. J. Am. Acad. Dermatol. 2014, 71, 185–191. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Diseases | Clinical Features | Autoantigens [3] | Diagnosis [3,4] |
|---|---|---|---|
| Pemphigus vulgaris (PV) |
| Dsg1,3 |
|
| Pemphigus foliaceus (PF) |
| Dsg1 |
|
| Pemphigus erythematosus (PE) | Blisters on the erythematous plaques at the nose, nasolabial folds and malar regions [7] | Dsg1 |
|
| IgA pemphigus | Vesiculopustular lesions on the erythematous plaques in an annular morphology at the trunk and proximal extremities [8] | Dsc1,2,3 & Dsg1,3 |
|
| Paraneoplastic pemphigus (PNP) |
| Envoplakin, periplakin, BP230, Dsg1,3, desmoplakins, epiplakin, α-2- macroglobulin- like antigen-1, and plectin |
|
| Diseases | Clinical Features | Autoantigens [3] | Diagnosis [3,4] |
|---|---|---|---|
| Bullous pemphigoid (BP) |
| BP180, BP230 |
|
| Mucous membrane pemphigoid (MMP) | Blistering occur predominantly on the oral cavity and conjunctiva which frequently healed with scarring [16,17,18] | BP180, BP230, laminin 332, α6β4 integrin |
|
| Linear IgA bullous dermatosis |
| BP180 (LAD-1), type VII collagen |
|
| Epidermolysis bullosa acquisita (EBA) |
| Type VII collagen |
|
| Dermatitis herpetiformis (DH) |
| Epidermal/Tissue transglutaminase, endomysium, deamidated gliadin |
|
| Laminin γ1 pemphigoid |
| p200 protein, laminin γ1 |
|
| Disease | First-Line Treatment |
|---|---|
| Intraepithelial AIBDs | |
| Pemphigus vulgaris | High dose systemic glucocorticoids (0.5 to 1.5 mg/kg/day prednisolone) and rituximab (either the lymphoma (weekly dosage of 375 mg/m2 for four consecutive weeks) or the rheumatoid arthritis (2 doses of 1000 mg separated by 2 weeks; may be repeated 6 months later) protocol) |
| First-line adjuvants: Azathioprine (1 to 3 mg/kg/day), MMF (2 g/day) or mycophenolic acid (1440 mg/day) | |
| Pemphigus foliaceus | The same as PV but usually with lower dosage |
| Pemphigus erythematosus | Systemic glucocorticoids (0.5 to 1 mg/kg/day prednisolone) and dapsone (100 to 200 mg/day) |
| IgA pemphigus | Systemic glucocorticoids (0.5 to 1 mg/kg/day prednisolone) and dapsone (100 to 300 mg/day) |
| Paraneoplastic pemphigus | Control of the underlying malignancy, systemic prednisolone (0.5 to 1 mg/kg/day) and rituximab (either the lymphoma (weekly dosage of 375 mg/m2 for four consecutive weeks) or the rheumatoid arthritis (2 doses of 1000 mg separated by 2 weeks; may be repeated 6 months later) protocol) |
| Subepithelial AIBDs | |
| Bullous pemphigoid | Systemic glucocorticoids (0.5 to 0.75 mg/kg/day of prednisolone) or high potency topical glucocorticoids, and tetracyclines ± niacinamide |
| Mucous membrane pemphigoid | Systemic glucocorticoids (0.25 to 0.5 mg/kg/day prednisolone) and dapsone (50 to 200 mg/day) |
| Linear IgA bullous dermatosis | Topical high potency glucocorticoids and dapsone (~2 mg/kg/day for children; 100 to 200 mg/day for adults) |
| Epidermolysis bullosa acquisita | High dose systemic glucocorticoids (1 to 1.5 mg/kg/day), dapsone (25 to 100 mg/day) and colchicine (0.6 to 1.2 mg/day) |
| Dermatitis herpetiformis | Gluten-free diet and dapsone (50 to 150 mg/day) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, K.-Y.; Yu, H.-S.; Yu, S. Current and Innovated Managements for Autoimmune Bullous Skin Disorders: An Overview. J. Clin. Med. 2022, 11, 3528. https://doi.org/10.3390/jcm11123528
Chu K-Y, Yu H-S, Yu S. Current and Innovated Managements for Autoimmune Bullous Skin Disorders: An Overview. Journal of Clinical Medicine. 2022; 11(12):3528. https://doi.org/10.3390/jcm11123528
Chicago/Turabian StyleChu, Kuan-Yu, Hsin-Su Yu, and Sebastian Yu. 2022. "Current and Innovated Managements for Autoimmune Bullous Skin Disorders: An Overview" Journal of Clinical Medicine 11, no. 12: 3528. https://doi.org/10.3390/jcm11123528