
Lethal Congenital Contracture Syndrome 11: A Case Report and Literature Review

 ,
,  , , , , , , and
, , , , , , and
Abstract
:1. Introduction
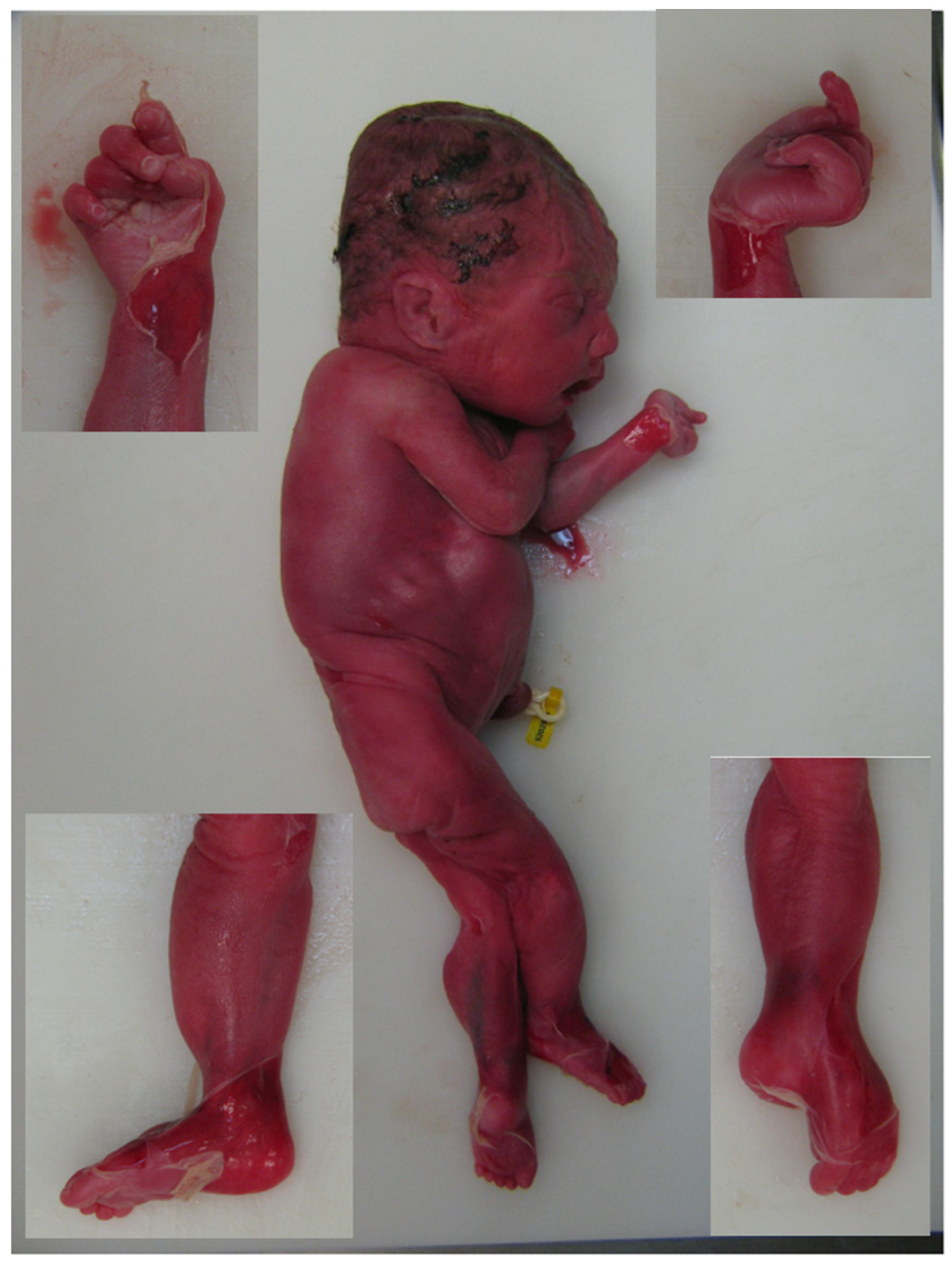
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hall, J.G.; Kimber, E.; Dieterich, K. Classification of arthrogryposis. Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Lowry, R.B.; Sibbald, B.; Bedard, T.; Hall, J.G. Prevalence of multiple congenital contractures including arthrogryposis multiplex congenita in Alberta, Canada, and a strategy for classification and coding. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.; Hall, J.G. Gene ontology analysis of arthrogryposis (multiple congenital contractures). Am. J. Med. Genet. C Semin. Med. Genet. 2019, 181, 310–326. [Google Scholar] [CrossRef]
- Niles, K.M.; Blaser, S.; Shannon, P.; Chitayat, D. Fetal arthrogryposis multiplex congenita/fetal akinesia deformation sequence (FADS)-Aetiology, diagnosis, and management. Prenat. Diagn. 2019, 39, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Skaria, P.; Dahl, A.; Ahmed, A. Arthrogryposis multiplex congenita in utero: Radiologic and pathologic findings. J. Matern. Fetal Neonatal Med. 2019, 32, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Ravenscroft, G.; Clayton, J.S.; Faiz, F.; Sivadorai, P.; Milnes, D.; Cincotta, R.; Moon, P.; Kamien, B.; Edwards, M.; Delatycki, M.; et al. Neurogenetic fetal akinesia and arthrogryposis: Genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 2021, 58, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Pergande, M.; Motameny, S.; Özdemir, Ö.; Kreutzer, M.; Wang, H.; Daimagüler, H.S.; Becker, K.; Karakaya, M.; Ehrhardt, H.; Elcioglu, N.; et al. The genomic and clinical landscape of fetal akinesia. Genet. Med. 2020, 22, 511–523. [Google Scholar] [CrossRef]
- Laquerriere, A.; Jaber, D.; Abiusi, E.; Maluenda, J.; Mejlachowicz, D.; Vivanti, A.; Dieterich, K.; Stoeva, R.; Quevarec, L.; Nolent, F.; et al. Phenotypic spectrum and genomics of undiagnosed arthrogryposis multiplex congenita. J. Med. Genet. 2021, 59, 559–567. [Google Scholar] [CrossRef]
- Maluenda, J.; Manso, C.; Quevarec, L.; Vivanti, A.; Marguet, F.; Gonzales, M.; Guimiot, F.; Petit, F.; Toutain, A.; Whalen, S.; et al. Mutations in GLDN, Encoding Gliomedin, a Critical Component of the Nodes of Ranvier, Are Responsible for Lethal Arthrogryposis. Am. J. Hum. Genet. 2016, 99, 928–933. [Google Scholar] [CrossRef] [Green Version]
- Wambach, J.A.; Stettner, G.M.; Haack, T.B.; Writzl, K.; Škofljanec, A.; Maver, A.; Munell, F.; Ossowski, S.; Bosio, M.; Wegner, D.J.; et al. Survival among children with “Lethal” congenital contracture syndrome 11 caused by novel mutations in the gliomedin gene (GLDN). Hum. Mutat. 2017, 38, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.W.; Murrell, J.R.; Nesbitt, A.I.; Pechter, K.B.; Balciuniene, J.; Zhao, X.; Yu, Z.; Denenberg, E.H.; DeChene, E.T.; Wilkens, A.B.; et al. Automated Clinical Exome Reanalysis Reveals Novel Diagnoses. J. Mol. Diagn. 2019, 21, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Lai, Y.; Yan, Z.; Wang, Y.; Nie, Y.; Guan, S.; Kuo, Y.; Zhang, W.; Zhu, X.; Peng, M.; et al. Trio-exome sequencing and preimplantation genetic diagnosis for unexplained recurrent fetal malformations. Hum. Mutat. 2020, 41, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Mis, E.K.; Al-Ali, S.; Ji, W.; Spencer-Manzon, M.; Konstantino, M.; Khokha, M.K.; Jeffries, L.; Lakhani, S.A. The latest FADS: Functional analysis of GLDN patient variants and classification of GLDN-associated AMC as a type of viable fetal akinesia deformation sequence. Am. J. Med. Genet. A 2020, 182, 2291–2296. [Google Scholar] [CrossRef] [PubMed]
- Australian Genomics Health Alliance Acute Care Flagship; Lunke, S.; Eggers, S.; Wilson, M.; Patel, C.; Barnett, C.P.; Pinner, J.; Sandaradura, S.A.; Buckley, M.F.; Krzesinski, E.I.; et al. Feasibility of Ultra-Rapid Exome Sequencing in Critically Ill Infants and Children With Suspected Monogenic Conditions in the Australian Public Health Care System. JAMA 2020, 323, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Reischer, T.; Liebmann-Reindl, S.; Bettelheim, D.; Balendran-Braun, S.; Streubel, B. Genetic diagnosis and clinical evaluation of severe fetal akinesia syndrome. Prenat. Diagn. 2020, 40, 1532–1539. [Google Scholar] [CrossRef]
- Bertoli-Avella, A.M.; Beetz, C.; Ameziane, N.; Rocha, M.E.; Guatibonza, P.; Pereira, C.; Calvo, M.; Herrera-Ordonez, N.; Segura-Castel, M.; Diego-Alvarez, D.; et al. Successful application of genome sequencing in a diagnostic setting: 1007 index cases from a clinically heterogeneous cohort. Eur. J. Hum. Genet. 2021, 29, 141–153. [Google Scholar] [CrossRef]
- Filges, I.; Hall, J.G. Failure to identify antenatal multiple congenital contractures and fetal akinesia–proposal of guidelines to improve diagnosis. Prenat. Diagn. 2013, 33, 61–74. [Google Scholar] [CrossRef]
- Tjon, J.K.; Tan-Sindhunata, M.B.; Bugiani, M.; Witbreuk, M.M.E.H.; van der Sluijs, J.A.; Weiss, M.M.; van Weissenbruch, M.M.; van de Pol, L.A.; Buizer, A.I.; van Doesburg, M.H.M.; et al. Care Pathway for Foetal Joint Contractures, Foetal Akinesia Deformation Sequence, and Arthrogryposis Multiplex Congenita. Fetal Diagn. Ther. 2021, 48, 829–839. [Google Scholar] [CrossRef]
- Eshed, Y.; Feinberg, K.; Poliak, S.; Sabanay, H.; Sarig-Nadir, O.; Spiegel, I.; Bermingham, J.R., Jr.; Peles, E. Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 2005, 47, 215–229. [Google Scholar] [CrossRef] [Green Version]
- Eshed, Y.; Feinberg, K.; Carey, D.J.; Peles, E. Secreted gliomedin is a perinodal matrix component of peripheral nerves. J. Cell Biol. 2007, 177, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Maertens, B.; Hopkins, D.; Franzke, C.W.; Keene, D.R.; Bruckner-Tuderman, L.; Greenspan, D.S.; Koch, M. Cleavage and oligomerization of gliomedin, a transmembrane collagen required for node of ranvier formation. J. Biol. Chem. 2007, 282, 10647–10659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labasque, M.; Devaux, J.J.; Lévêque, C.; Faivre-Sarrailh, C. Fibronectin type III-like domains of neurofascin-186 protein mediate gliomedin binding and its clustering at the developing nodes of Ranvier. J. Biol. Chem. 2011, 286, 42426–42434. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Prenatal Ultrasound Examination | Fetal Death | Postmortem Examination | Birth | Genetic Variant 1 | Genetic Variant 2 | Reference |
|---|---|---|---|---|---|---|---|---|
| Family 1 Case 1 | male | 32 wg: Akinesia Polyhydramnios | Exitus 33 wg | Extension of lower limbs Extension contractures of wrists Pulmonary hypoplasia | - | c.758delC p.(Pro253LeufsTer51) | c.1423G>C p.(Ala475Pro) | [9] |
| Family 1 Case 2 | female | Akinesia Polyhydramnios | TOP 33 wg | Unremarkable histological examination of the spinal cord and skeletal muscle Reduced number of myelinated fibers | - | c.758delC p.(Pro253LeufsTer51) | c.1423G>C p.(Ala475Pro) | [9] |
| Family 2 Case 1 | male | 30 wg Polyhydramnios Intrauterine growth retardation AMC (flexion contractures of the elbows, extension of the knees, camptodactyly, and retrognathia) | - | NI | 30 wg AMC (flexion contractures of the elbows, extension of the knees, camptodactyly, and retrognathia) Exitus: day 1 | c.95C>A p.(Ala32Glu) | c.95C>A p.(Ala32Glu) | [9] |
| Family 3 Case 1 | male | 28 wg: Akinesia Polyhydramnios Bilateral flexion of fingers | - | Unremarkable pathological examination of the brain and spinal cord | AMC (involving the fingers, wrists, thumbs, and knees) Pulmonary hypoplasia Exitus: day 1 | c.541 + 1G>A | c.1240C>T p.(Arg414Ter) | [9] |
| Family 3 Case 2 | male | 31 wg: Polyhydramnios Bilateral flexion of fingers Reduced mobility | TOP 31 wg | AMC with microretrognathia Pulmonary hypoplasia | - | c.541 + 1G>A | c.1240C>T p.(Arg414Ter) | [9] |
| Family 4 Case 1 | female | 27 wg: Reduced mobility Polyhydramnios 29 wg: Fetal Immobility | TOP 30 wg | Unremarkable pathological examination of the brain and spinal cord | Distal arthrogryposis of the hands Bilateral club foot Pulmonary hypoplasia | c.1435C>T p.(Arg479Ter) | c.1435C>T p.(Arg479Ter) | [9] |
| Family 5 Case 1 | male | Reduced mobility Breech | - | AMC Pulmonary hypoplasia and pulmonary hemorrhage Bilateral hip dislocations Fistula from the left anterior descending artery to right ventricle Bilateral small kidneys with calcifications, an ectopic right ureter without signs of obstruction, and intraventricular hemorrhage Skeletal muscle fibers were small for age and central nuclei suggested centronuclear myopathy | 38 wg Respiratory failure Exitus: day 2 | c.927_930del p.(Asn309LysfsTer5) | c.1436G>C p.(Arg479Pro) | [10] |
| Family 5 Case 2 | female | Polyhydramnios Intrauterine growth restriction Bilateral club feet | - | - | 37 wg Respiratory insufficiency Contractures of hips, knees fixed in extension Bilateral club feet Flexion contracture of left long finger Bilateral hip dislocation Axial and appendicular hypotonia Alive at 22 months with tracheostomy and home ventilation | c.927_930del p.(Asn309LysfsTer5) | c.1436G>C p.(Arg479Pro) | [10] |
| Family 5 Case 3 | male | Polyhydramnios Bilateral club feet Flexed wrists Extended knees Breech Intrauterine growth restriction | - | - | 39 wg Respiratory insufficiency Contractures of hips, knees Bilateral club feet Hyperextension of thumbs to radii Axial and appendicular hypotonia Undescended testes Alive at 7 months with tracheostomy and home ventilation | c.927_930del p.(Asn309LysfsTer5) | c.1436G>C p.(Arg479Pro) | [10] |
| Family 6 Case 1 | male | Polyhydramnios | - | - | 33 wg Pulmonary hypoplasia Bilateral hip dislocation Contractures of knees and wrists Bilateral club feet Progressive scoliosis, diaphragm paralysis, borderline intellectual functioning (IQ 74) Alive at age 17 years old with intermittent use of non-invasive mask ventilation | c.1305G>A p.(Trp435Ter) | c.1305G>A p.(Trp435Ter) | [10] |
| Family 7 Case 1 | female | 30 wg. Akinesia Polyhidramnios Skin edema | TOP 31 wg | NI | - | c.1305G>A p.(Trp435Ter) | c.1305G>A p.(Trp435Ter) | [10] |
| Family 7 Case 2 | male | - | - | - | 41 wg Paresis of right vocal cord and right side of the soft palate Bilateral hip flexion contractures with dislocated hips Extension contractures of kneesCalcaneovalgus deformity of feet Axial and appendicular hypotonia Atrophy of lower limbs Right-sided cryptorchidism Intubated at birth for respiratory failure Tracheostomy at 6 weeks of age Alive at 28 months without ventilatory support | c.1305G>A p.(Trp435Ter) | c.1305G>A p.(Trp435Ter) | [10] |
| Family 8 Case 1 | male | Akinesia Flexed arms and closed hand | TOP 27 wg | Pulmonary hypoplasia Extension contractures of hip sand knees Flexion contractures of fingers | - | Unknown | Unknown | [10] |
| Family 8 Case 2 | female | 26 wg: Polyhydramnios Arthrogryposis | - | - | 36 wg: Pulmonary hypoplasia Extension contractures of hips and knees Flexion contractures of elbows, wrists, and fingers Bilateral vertical talus information Diffuse muscle atrophy/hypoplasia Exitus: 12 h | c.1178G>A p.(Arg393Lys) | c.1428C>A p.(Phe476Leu) | [10] |
| Family 9 Case 1 | male | 26 wg: Multiple joint contracture Polyhydramnios | - | - | - | c.1027G>A p.(Gly343Ser) | c.1240C>T p.(Arg414Ter) | [13] |
| Family 9 Case 2 | female | 26 wg: Multiple joint contracture Polyhydramnios | - | - | - | c.1027G>A p.(Gly343Ser) | c.1240C>T p.(Arg414Ter) | [13] |
| Family 10 Case 1 | - | NI | NI | NI | NI | c.1494G>C p.(Leu498Phe) | c.1494G>C p.(Leu498Phe) | [12] |
| Family 11 Case 1 | female | Early fetal demise of a twin <12 wg Polyhydramnios Preterm premature rupture of membranes Breech (20 wg) | - | - | 30 wg: Bilateral extension knee contractures and camptodactyly Bilateral congenital hip dysplasia and right-sided hip dislocation Hypotonia Pulmonary hypoplasia Alive at 44 months | c.1093C>T p.(Leu365Phe) | c.1178G>A p.(Arg393Lys) | [14] |
| Family 12 Case 1 | female | Fetal akinesia | NI | NI | Joint contractures: Hips, knees, ankles, elbows, fingers Microcephaly Delayed motor development Muscular hypertonia Hip joint luxation Alive at 1 year | c.1178G>A p.(Arg393Lys) | c.1428C>A p.(Phe476Leu) | [7] |
| Family 13 Case 1 | male | Hydrops fetalis | - | - | Subtle joint contractures Down-slanted palpebral fissures Ventilator support Care redirected towards palliation | c.980_981del p.(Ser327CysfsTer2) | c.980_981del p.(Ser327CysfsTer2) | [15] |
| Family 14 Case 1 | male | No findings | - | - | Exitus: < 1 month | c.95C>A p.(Ala32Glu) | c.95C>A p.(Ala32Glu) | [8] * |
| Family 15 Case 1 | female | Abnormalities | TOP | NI | c.1435C>T p.(Arg479Ter) | c.1435C>T p.(Arg479Ter) | [8] * | |
| Family 16 Case 1 + Case 2 | Female (2 cases) | Abnormalities | - | NI | Exitus: 2 months | c.82G>C p.(Ala28Pro) | c.1241G>A p.(Arg414Gln) | [8] * |
| Family 17 Case 1 | - | 32 wg: Polyhydramnios Missing fetal movements Facial dismorphism Lung hypoplasia Flexed knees, extended anckles, flexed elbows, fisted hands | - | - | 32 wg Exitus: 1 day | c.1423G>C p.(Ala475Pro) | c.1423G>C p.(Ala475Pro) | [16] |
| Family 17 Case 2 | - | 23 wg: Polyhydramnios Missing fetal movements Microcephaly Single umbilical artery Pericardial and pleural effusion Flexed knees, flexed elbows, fisted hands | TOP 27 wg | - | - | c.1423G>C p.(Ala475Pro) | c.1423G>C p.(Ala475Pro) | [16] |
| Family 18 Case 1 | - | NI | - | - | Flexion contracture Hydrops fetalis Pulmonary hypoplasia Pleural effusion | c.1028-2A>T | c.1028-2A>T | [17] |
| PRESENT CASE | female | 28 wg: Hydrops fetalis Arthrogryposis | TOP 29 wg | Distal arthrogryposis of the hands Left club foot Pulmonary hypoplasia Retrognathia | - | c.62C>A p.(Ala21Glu) | c.1494G > T p.(Leu498Phe) | PRESENT STUDY |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potrony, M.; Borrell, A.; Masoller, N.; Nadal, A.; Rodriguez-Carunchio, L.; Saez de Gordoa Elizalde, K.; Quesada-Espinosa, J.F.; Villanueva-Cañas, J.L.; Pauta, M.; Jodar, M.; et al. Lethal Congenital Contracture Syndrome 11: A Case Report and Literature Review. J. Clin. Med. 2022, 11, 3570. https://doi.org/10.3390/jcm11133570
Potrony M, Borrell A, Masoller N, Nadal A, Rodriguez-Carunchio L, Saez de Gordoa Elizalde K, Quesada-Espinosa JF, Villanueva-Cañas JL, Pauta M, Jodar M, et al. Lethal Congenital Contracture Syndrome 11: A Case Report and Literature Review. Journal of Clinical Medicine. 2022; 11(13):3570. https://doi.org/10.3390/jcm11133570
Chicago/Turabian StylePotrony, Miriam, Antoni Borrell, Narcís Masoller, Alfons Nadal, Leonardo Rodriguez-Carunchio, Karmele Saez de Gordoa Elizalde, Juan Francisco Quesada-Espinosa, Jose Luis Villanueva-Cañas, Montse Pauta, Meritxell Jodar, and et al. 2022. "Lethal Congenital Contracture Syndrome 11: A Case Report and Literature Review" Journal of Clinical Medicine 11, no. 13: 3570. https://doi.org/10.3390/jcm11133570
APA StylePotrony, M., Borrell, A., Masoller, N., Nadal, A., Rodriguez-Carunchio, L., Saez de Gordoa Elizalde, K., Quesada-Espinosa, J. F., Villanueva-Cañas, J. L., Pauta, M., Jodar, M., Madrigal, I., Badenas, C., Alvarez-Mora, M. I., & Rodriguez-Revenga, L. (2022). Lethal Congenital Contracture Syndrome 11: A Case Report and Literature Review. Journal of Clinical Medicine, 11(13), 3570. https://doi.org/10.3390/jcm11133570