
Identification of Genetic Variations in the NAD-Related Pathways for Patients with Major Depressive Disorder: A Case-Control Study in Taiwan

 ,
,  , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
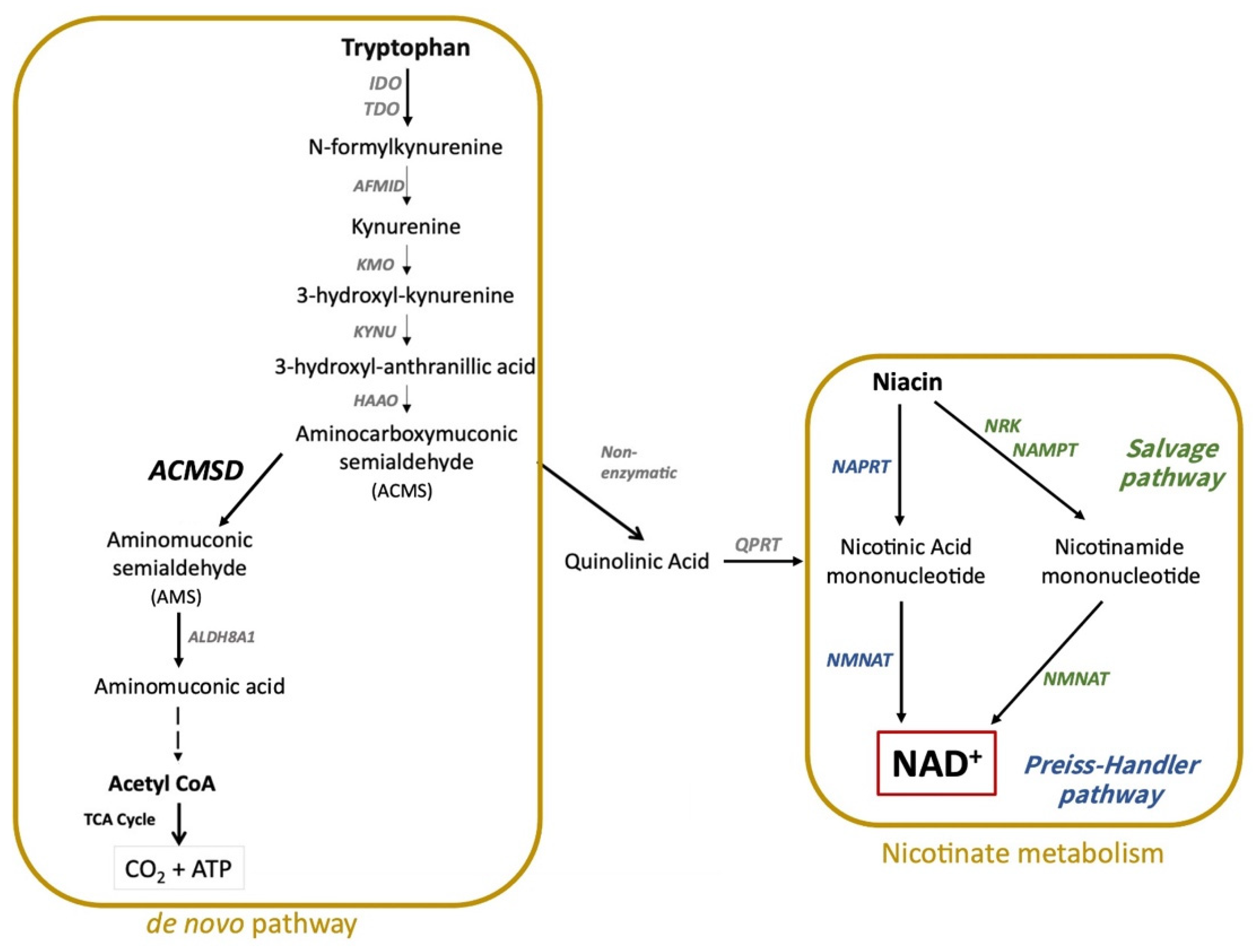
2.1. Participants Selection and Cohort Building
2.2. Genotyping
2.3. Data Quality Control
2.4. Statistical Analysis
3. Results
3.1. Candidate Gene Selection
3.2. Full-Model Case-Control Association Test
3.3. Logistic Regression
3.4. Haplotype Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sultani, G.; Samsudeen, A.F.; Osborne, B.; Turner, N. NAD(+): A key metabolic regulator with great therapeutic potential. J. Neuroendocrinol. 2017, 29, e12508. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Sauve, A.A. NAD(+) metabolism: Bioenergetics, signaling and manipulation for therapy. Biochim. Biophys. Acta 2016, 1864, 1787–1800. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J. ACMSD: A Novel Target for Modulating NAD(+) Homeostasis. Trends Endocrinol. Metab. 2019, 30, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Li, Y.; Li, Y.; Wang, X. Molecular properties, functions, and potential applications of NAD kinases. Acta Biochim. Biophys. Sin. 2009, 41, 352–361. [Google Scholar] [CrossRef]
- Lam, N.S.K.; Long, X.X.; Li, X.; Saad, M.; Lim, F.; Doery, J.C.; Griffin, R.C.; Galletly, C. The potential use of folate and its derivatives in treating psychiatric disorders: A systematic review. Biomed. Pharmacother. 2022, 146, 112541. [Google Scholar] [CrossRef]
- Wang, B.; Lian, Y.J.; Su, W.J.; Peng, W.; Dong, X.; Liu, L.L.; Gong, H.; Zhang, T.; Jiang, C.L.; Wang, Y.X. HMGB1 mediates depressive behavior induced by chronic stress through activating the kynurenine pathway. Brain Behav. Immun. 2018, 72, 51–60. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef]
- Arnau-Soler, A.; Macdonald-Dunlop, E.; Adams, M.J.; Clarke, T.K.; MacIntyre, D.J.; Milburn, K.; Navrady, L.; Hayward, C.; McIntosh, A.M.; Thomson, P.A. Genome-wide by environment interaction studies of depressive symptoms and psychosocial stress in UK Biobank and Generation Scotland. Transl. Psychiatry 2019, 9, 14. [Google Scholar] [CrossRef]
- Liu, W.; Yan, H.; Zhou, D.; Cai, X.; Zhang, Y.; Li, S.; Li, H.; Li, S.; Zhou, D.S.; Li, X.; et al. The depression GWAS risk allele predicts smaller cerebellar gray matter volume and reduced SIRT1 mRNA expression in Chinese population. Transl. Psychiatry 2019, 9, 333. [Google Scholar] [CrossRef]
- Cheng, S.W.; Li, J.X.; Chen, D.T.; Chien, Y.C.; Chang, J.P.; Huang, S.Y.; Galecki, P.; Su, K.P. Predictive Genetic Variations in the Kynurenine Pathway for Interferon-alpha-Induced Depression in Patients with Hepatitis C Viral Infection. J. Pers. Med. 2021, 11, 192. [Google Scholar] [CrossRef]
- Cheng, S.W.; Li, J.X.; Chien, Y.C.; Chang, J.P.; Shityakov, S.; Huang, S.Y.; Galecki, P.; Su, K.P. Genetic Variations of Ionotropic Glutamate Receptor Pathways on Interferon-alpha-induced Depression in Patients with Hepatitis C Viral Infection. Brain Behav. Immun. 2021, 93, 16–22. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Kennedy, S.H. Core symptoms of major depressive disorder: Relevance to diagnosis and treatment. Dialogues Clin. Neurosci. 2008, 10, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Hasin, D.S.; Sarvet, A.L.; Meyers, J.L.; Saha, T.D.; Ruan, W.J.; Stohl, M.; Grant, B.F. Epidemiology of Adult DSM-5 Major Depressive Disorder and Its Specifiers in the United States. JAMA Psychiatry 2018, 75, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Lewinsohn, P.M.; Clarke, G.N.; Seeley, J.R.; Rohde, P. Major depression in community adolescents: Age at onset, episode duration, and time to recurrence. J. Am. Acad. Child Adolesc. Psychiatry 1994, 33, 809–818. [Google Scholar] [CrossRef]
- Gaynes, B.N.; Warden, D.; Trivedi, M.H.; Wisniewski, S.R.; Fava, M.; Rush, A.J. What did STAR*D teach us? Results from a large-scale, practical, clinical trial for patients with depression. Psychiatr. Serv. 2009, 60, 1439–1445. [Google Scholar] [CrossRef]
- Nemeroff, C.B. Prevalence and management of treatment-resistant depression. J. Clin. Psychiatry 2007, 68 (Suppl. S8), 17–25. [Google Scholar]
- Imai, S.I.; Guarente, L. It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2016, 2, 16017. [Google Scholar] [CrossRef]
- O’Callaghan, C.; Vassilopoulos, A. Sirtuins at the crossroads of stemness, aging, and cancer. Aging Cell 2017, 16, 1208–1218. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD(+) metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef]
- Asher, G.; Sassone-Corsi, P. Time for food: The intimate interplay between nutrition, metabolism, and the circadian clock. Cell 2015, 161, 84–92. [Google Scholar] [CrossRef]
- Paulionis, L.; Kane, S.L.; Meckling, K.A. Vitamin status and cognitive function in a long-term care population. BMC Geriatr. 2005, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Cohen, B.M.; Chen, X.; Lukas, S.E.; Shinn, A.K.; Yuksel, A.C.; Li, T.; Du, F.; Öngür, D. Redox Dysregulation in Schizophrenia Revealed by in vivo NAD+/NADH Measurement. Schizophr. Bull. 2016, 43, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Hollis, F.; van der Kooij, M.A.; Zanoletti, O.; Lozano, L.; Cantó, C.; Sandi, C. Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. USA 2015, 112, 15486–15491. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, B.H. Nicotinic Acid Long-Term Effectiveness in a Patient with Bipolar Type II Disorder: A Case of Vitamin Dependency. Nutrients 2018, 10, 134. [Google Scholar] [CrossRef]
- Viljoen, M.; Swanepoel, A.; Bipath, P. Antidepressants may lead to a decrease in niacin and NAD in patients with poor dietary intake. Med. Hypotheses 2015, 84, 178–182. [Google Scholar] [CrossRef]
- Anderson, G. Linking the biological underpinnings of depression: Role of mitochondria interactions with melatonin, inflammation, sirtuins, tryptophan catabolites, DNA repair and oxidative and nitrosative stress, with consequences for classification and cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 255–266. [Google Scholar] [CrossRef]
- Mithaiwala, M.N.; Santana-Coelho, D.; Porter, G.A.; O’Connor, J.C. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells 2021, 10, 1548. [Google Scholar] [CrossRef]
- Palzer, L.; Bader, J.J.; Angel, F.; Witzel, M.; Blaser, S.; McNeil, A.; Wandersee, M.K.; Leu, N.A.; Lengner, C.J.; Cho, C.E.; et al. Alpha-Amino-Beta-Carboxy-Muconate-Semialdehyde Decarboxylase Controls Dietary Niacin Requirements for NAD(+) Synthesis. Cell Rep. 2018, 25, 1359–1370.e4. [Google Scholar] [CrossRef]
- Liu, F.; Dong, Y.Y.; Lei, G.; Zhou, Y.; Liu, P.; Dang, Y.H. HINT1 Is Involved in the Chronic Mild Stress Elicited Oxidative Stress and Apoptosis Through the PKC epsilon/ALDH-2/4HNE Pathway in Prefrontal Cortex of Rats. Front. Behav. Neurosci. 2021, 15, 690344. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Merikangas, K.R.; Walters, E.E. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005, 62, 593–602. [Google Scholar] [CrossRef]
- Wigginton, J.E.; Cutler, D.J.; Abecasis, G.R. A note on exact tests of hardy-weinberg equilibrium. Am. J. Hum. Genet. 2005, 76, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.S.; Niu, T.; Liu, J.S. Partition-ligation–expectation-maximization algorithm for haplotype inference with single-nucleotide polymorphisms. Am. J. Hum. Genet. 2002, 71, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Jack, J.R.; Motsinger-Reif, A.A.; Brown, C.C. An adaptive permutation approach for genome-wide association study: Evaluation and recommendations for use. BioData Min. 2014, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef]
- Liu, H.M.; Zheng, J.P.; Yang, D.; Liu, Z.F.; Li, Z.; Hu, Z.Z.; Li, Z.N. Recessive/dominant model: Alternative choice in case-control-based genome-wide association studies. PLoS ONE 2021, 16, e0254947. [Google Scholar] [CrossRef]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; de Vries, J.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Primers 2021, 1, 59. [Google Scholar] [CrossRef]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]
- Rose, A.B. Intron-mediated regulation of gene expression. Curr. Top Microbiol. Immunol. 2008, 326, 277–290. [Google Scholar] [CrossRef]
- Rose, A.B. Introns as Gene Regulators: A Brick on the Accelerator. Front. Genet. 2018, 9, 672. [Google Scholar] [CrossRef]
- Chen, C.H.; Kraemer, B.R.; Lee, L.; Mochly-Rosen, D. Annotation of 1350 Common Genetic Variants of the 19 ALDH Multigene Family from Global Human Genome Aggregation Database (gnomAD). Biomolecules 2021, 11, 1423. [Google Scholar] [CrossRef] [PubMed]
- Krupenko, N.I.; Dubard, M.E.; Strickland, K.C.; Moxley, K.M.; Oleinik, N.V.; Krupenko, S.A. ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J. Biol. Chem. 2010, 285, 23056–23063. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.A.; Bell, S.J.; Guan, Y.; Yu, Y.H. Folic Acid supplementation and pregnancy: More than just neural tube defect prevention. Rev. Obstet. Gynecol. 2011, 4, 52–59. [Google Scholar] [PubMed]
- Lewis, S.J.; Araya, R.; Leary, S.; Smith, G.D.; Ness, A. Folic acid supplementation during pregnancy may protect against depression 21 months after pregnancy, an effect modified by MTHFR C677T genotype. Eur. J. Clin. Nutr. 2012, 66, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Liu, Y.; Cao, L.; Zheng, Y.; Li, W.; Huang, G. Association between Duration of Folic Acid Supplementation during Pregnancy and Risk of Postpartum Depression. Nutrients 2017, 9, 1206. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Campo, P.M.; Almeida, J.; Sanchez, M.L.; Malvezzi, M.; Orfao, A. Normal patterns of expression of glycosylphosphatidylinositol-anchored proteins on different subsets of peripheral blood cells: A frame of reference for the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytom. B Clin. Cytom. 2006, 70, 71–81. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Ferracin, M.; Ghimenti, C.; Massaia, M.; Horenstein, A.L.; Malavasi, F. Anti-CD38 antibody therapy: Windows of opportunity yielded by the functional characteristics of the target molecule. Mol. Med. 2013, 19, 99–108. [Google Scholar] [CrossRef]
- Higashida, H.; Liang, M.; Yoshihara, T.; Akther, S.; Fakhrul, A.; Stanislav, C.; Nam, T.S.; Kim, U.H.; Kasai, S.; Nishimura, T.; et al. An immunohistochemical, enzymatic, and behavioral study of CD157/BST-1 as a neuroregulator. BMC Neurosci. 2017, 18, 35. [Google Scholar] [CrossRef]
- Lopatina, O.; Yoshihara, T.; Nishimura, T.; Zhong, J.; Akther, S.; Fakhrul, A.A.; Liang, M.; Higashida, C.; Sumi, K.; Furuhara, K.; et al. Anxiety- and depression-like behavior in mice lacking the CD157/BST1 gene, a risk factor for Parkinson’s disease. Front. Behav. Neurosci. 2014, 8, 133. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Yoshihara, T.; Lopatina, O.; Ishihara, K.; Higashida, H. Selegiline Ameliorates Depression-Like Behavior in Mice Lacking the CD157/BST1 Gene, a Risk Factor for Parkinson’s Disease. Front. Behav. Neurosci. 2017, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Radenkovic, D.; Reason; Verdin, E. Clinical Evidence for Targeting NAD Therapeutically. Pharmaceuticals 2020, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Tsilioni, I.; Taliou, A.; Francis, K.; Theoharides, T.C. Children with autism spectrum disorders, who improved with a luteolin-containing dietary formulation, show reduced serum levels of TNF and IL-6. Transl. Psychiatry 2015, 5, e647. [Google Scholar] [CrossRef] [PubMed]
- Rao, T.S.; Asha, M.; Ramesh, B.; Rao, K.J. Understanding nutrition, depression and mental illnesses. Indian J. Psychiatry 2008, 50, 77. [Google Scholar]
- Yirmiya, R.; Weidenfeld, J.; Pollak, Y.; Morag, M.; Morag, A.; Avitsur, R.; Barak, O.; Reichenberg, A.; Cohen, E.; Shavit, Y. Cytokines, “depression due to a general medical condition,” and antidepressant drugs. In Cytokines, Stress, and Depression; Springer: New York, NY, USA, 1999; pp. 283–316. [Google Scholar]
- Leonard, B.E. Impact of inflammation on neurotransmitter changes in major depression: An insight into the action of antidepressants. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 48, 261–267. [Google Scholar] [CrossRef]
- Hashioka, S.; McGeer, P.L.; Monji, A.; Kanba, S. Anti-inflammatory effects of antidepressants: Possibilities for preventives against Alzheimer’s disease. Cent. Nerv. Syst. Agents Med. Chem. 2009, 9, 12–19. [Google Scholar] [CrossRef]
- Gao, X.; Becker, L.C.; Becker, D.M.; Starmer, J.D.; Province, M.A. Avoiding the high Bonferroni penalty in genome-wide association studies. Genet. Epidemiol. 2010, 34, 100–105. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Pathway | Official Symbol | Product Protein | Location (Chr) | Length (bp) | Number of SNPs after QC |
|---|---|---|---|---|---|
| KP | AADAT | Aminoadipate aminotransferase | 4 | 31,450 | 7 |
| KP | ACMSD | Aminocarboxymuconate semialdehyde decarboxylase | 2 | 63,610 | 10 |
| KP | AFMID | Arylformamidase | 17 | 20,384 | 3 |
| KP | HAAO | 3-hydroxyanthranilate 3,4-dioxygenase | 2 | 25,524 | 7 |
| KP | IDO1 | Indoleamine 2,3-dioxygenase 1 | 8 | 14,981 | 4 |
| KP | IDO2 | Indoleamine 2,3-dioxygenase 2 | 8 | 81,436 | 35 |
| KP | KMO | Kynurenine 3-monooxygenase | 1 | 63,625 | 33 |
| KP | KYAT1 | Kynurenine aminotransferase 1 | 9 | 48,962 | 4 |
| KP | KYAT3 | Kynurenine aminotransferase 3 | 1 | 57,187 | 5 |
| KP | KYNU | Kynureninase | 2 | 293,388 | 41 |
| KP | QPRT | Quinolinate phosphoribosyltransferase | 16 | 18,990 | 3 |
| KP | SLC36A4 | Solute carrier family 36 member 4 | 11 | 50,245 | 3 |
| KP | SLC3A2 | Solute carrier family 3 member 2 | 11 | 32,871 | 3 |
| KP | SLC7A5 | Solute carrier family 7 member 5 | 16 | 39,471 | 7 |
| KP | TDO2 | Tryptophan 2,3-dioxygenase | 4 | 16,713 | 1 |
| NM | BST1 | Bone marrow stromal cell antigen 1 | 4 | 29,722 | 14 |
| NM | CD38 | CD38 molecule | 4 | 74,904 | 16 |
| NM | NADK | NAD kinase | 1 | 28,799 | 2 |
| NM | NADK2 | NAD kinase 2, mitochondrial | 5 | 49,690 | 14 |
| NM | NADSYN1 | NAD synthetase 1 | 11 | 48,613 | 10 |
| NM | NMNAT1 | Nicotinamide nucleotide adenylyltransferase 1 | 1 | 42,575 | 6 |
| NM | NMNAT2 | Nicotinamide nucleotide adenylyltransferase 2 | 1 | 170,143 | 30 |
| NM | NMNAT3 | Nicotinamide nucleotide adenylyltransferase 3 | 3 | 117,870 | 18 |
| NM | NMRK1 | Nicotinamide riboside kinase 1 | 9 | 27,578 | 8 |
| NM | NMRK2 | Nicotinamide riboside kinase 2 | 19 | 9347 | 0 |
| NM | NT5E | 5’-nucleotidase ecto | 6 | 45,701 | 5 |
| NM | QPRT | Quinolinate phosphoribosyltransferase | 16 | 18,990 | 3 |
| Official Symbol | Product Protein | Location (Chr) | Length (bp) | Number of SNPs after QC |
|---|---|---|---|---|
| SIRT1 | Sirtuin 1 | 10 | 33,733 | 6 |
| SIRT2 | Sirtuin 2 | 19 | 21,063 | 3 |
| SIRT3 | Sirtuin 3 | 11 | 21,901 | 3 |
| SIRT4 | Sirtuin 4 | 12 | 21,469 | 1 |
| SIRT5 | Sirtuin 5 | 6 | 40,884 | 8 |
| SIRT6 | Sirtuin 6 | 19 | 8454 | 0 |
| SIRT7 | Sirtuin 7 | 17 | 6237 | 1 |
| ALDH1A1 | Aldehyde dehydrogenase 1 family member A1 | 9 | 52,382 | 15 |
| ALDH1A2 | Aldehyde dehydrogenase 1 family member A2 | 15 | 112,282 | 18 |
| ALDH1A3 | Aldehyde dehydrogenase 1 family member A3 | 15 | 3,6795 | 15 |
| ALDH1B1 | Aldehyde dehydrogenase 1 family member B1 | 9 | 5959 | 5 |
| ALDH1L1 | Aldehyde dehydrogenase 1 family member L1 | 3 | 77,616 | 25 |
| ALDH1L2 | Aldehyde dehydrogenase 1 family member L2 | 12 | 64,668 | 19 |
| ALDH2 | Aldehyde dehydrogenase 2 | 12 | 50,599 | 2 |
| ALDH3A1 | Aldehyde dehydrogenase 3 family member A1 | 17 | 10,314 | 9 |
| ALDH3A2 | Aldehyde dehydrogenase 3 family member A2 | 17 | 29,460 | 3 |
| ALDH3B1 | Aldehyde dehydrogenase 3 family member B1 | 11 | 20,726 | 6 |
| ALDH3B2 | Aldehyde dehydrogenase 3 family member B2 | 11 | 19,069 | 9 |
| ALDH4A1 | Aldehyde dehydrogenase 4 family member A1 | 1 | 31,125 | 4 |
| ALDH5A1 | Aldehyde dehydrogenase 5 family member A1 | 6 | 42,238 | 14 |
| ALDH6A1 | Aldehyde dehydrogenase 6 family member A1 | 14 | 27,606 | 6 |
| ALDH7A1 | Aldehyde dehydrogenase 7 family member A1 | 5 | 53,378 | 24 |
| ALDH8A1 | Aldehyde dehydrogenase 8 family member A1 | 6 | 32,708 | 5 |
| ALDH9A1 | Aldehyde dehydrogenase 9 family member A1 | 1 | 36451 | 6 |
| ALDH16A1 | Aldehyde dehydrogenase 16 family member A1 | 19 | 17,825 | 0 |
| ALDH18A1 | Aldehyde dehydrogenase 18 family member A1 | 10 | 50,770 | 9 |
| Pathway | Gene | SNP | Empirical p-Value | p-Value | Minor Allele | Major Allele | MAF in Cases | MAF in Controls |
|---|---|---|---|---|---|---|---|---|
| Tryptophan catabolism | ACMSD | rs12622574 | 0.0256 | 0.0035 | A | G | 0.1562 | 0.2078 |
| Nicotinate metabolism | BST1 | rs28532698 | 0.0476 | 0.0124 | C | T | 0.0883 | 0.0607 |
| CD38 | rs3733593 | 0.0230 | 0.0017 | A | G | 0.2437 | 0.3074 |
| Gene | SNP | Block Haplotype | Chi Square | p-Value | Permutation p-Value |
|---|---|---|---|---|---|
| ACMSD | rs12622574 | TTTACATT | 8.073 | 0.0045 | 0.0256 |
| BST1 | rs28532698 | GCGCC | 6.247 | 0.0124 | 0.1046 |
| GC | 7.887 | 0.0050 | 0.0476 | ||
| CD38 | rs3733593 | TCA | 9.347 | 0.0022 | 0.0230 |
| TCG | 6.122 | 0.0134 | 0.1060 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, D.T.-L.; Cheng, S.-W.; Chen, T.; Chang, J.P.-C.; Hwang, B.-F.; Chang, H.-H.; Chuang, E.Y.; Chen, C.-H.; Su, K.-P. Identification of Genetic Variations in the NAD-Related Pathways for Patients with Major Depressive Disorder: A Case-Control Study in Taiwan. J. Clin. Med. 2022, 11, 3622. https://doi.org/10.3390/jcm11133622
Chen DT-L, Cheng S-W, Chen T, Chang JP-C, Hwang B-F, Chang H-H, Chuang EY, Chen C-H, Su K-P. Identification of Genetic Variations in the NAD-Related Pathways for Patients with Major Depressive Disorder: A Case-Control Study in Taiwan. Journal of Clinical Medicine. 2022; 11(13):3622. https://doi.org/10.3390/jcm11133622
Chicago/Turabian StyleChen, Daniel Tzu-Li, Szu-Wei Cheng, Tiffany Chen, Jane Pei-Chen Chang, Bing-Fang Hwang, Hen-Hong Chang, Eric Y. Chuang, Che-Hong Chen, and Kuan-Pin Su. 2022. "Identification of Genetic Variations in the NAD-Related Pathways for Patients with Major Depressive Disorder: A Case-Control Study in Taiwan" Journal of Clinical Medicine 11, no. 13: 3622. https://doi.org/10.3390/jcm11133622