
Prenatal Diagnosis Using Chromosomal Microarray Analysis in High-Risk Pregnancies

 , and
, and 
Abstract
:1. Background
2. Materials and Methods
2.1. Patients and Indications
2.2. Prenatal Samples
2.3. DNA Preparation and Chromosomal Microarray Analysis
2.4. Interpretation and Reporting of CNVs
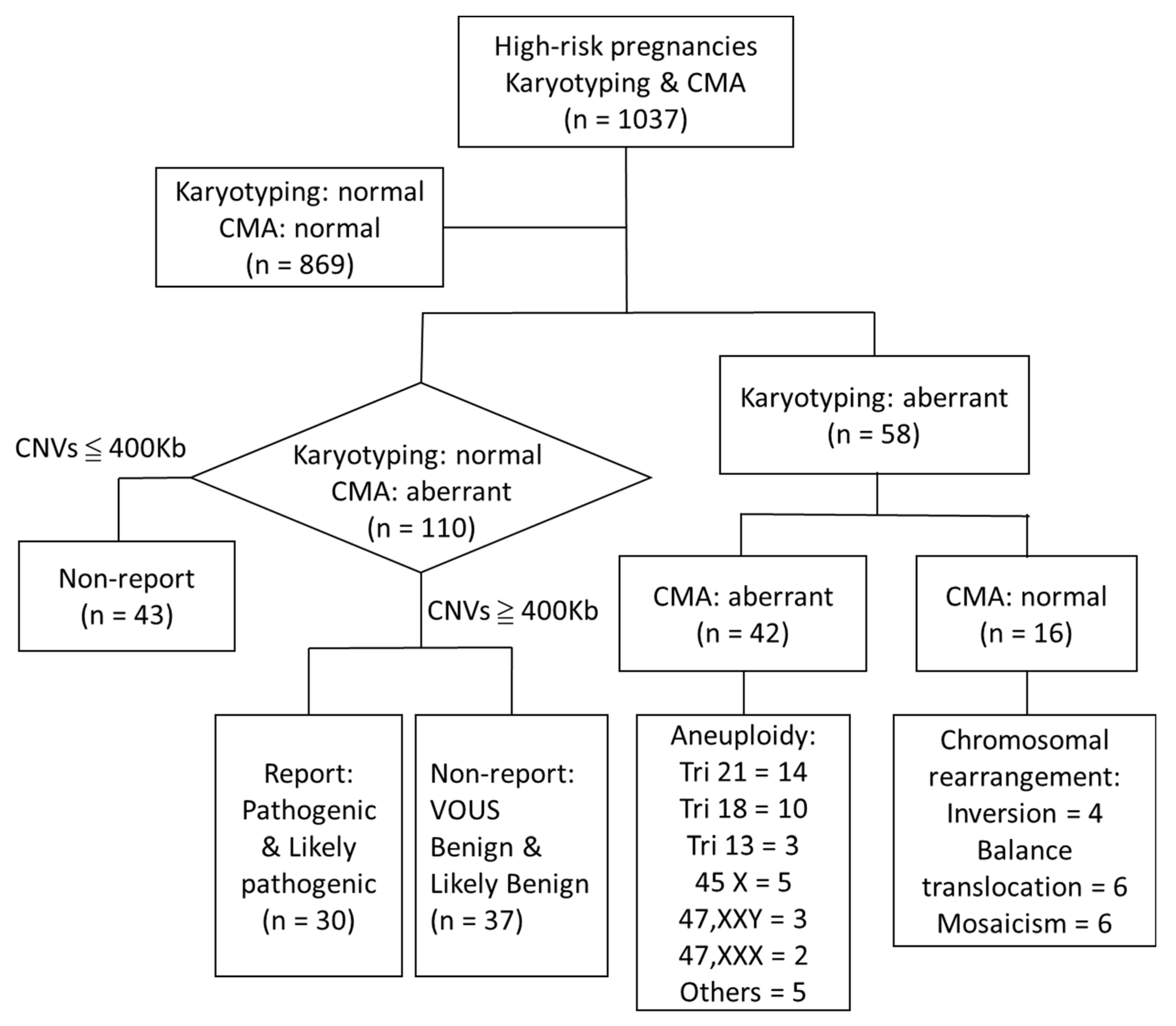
3. Results
4. Discussion
4.1. Overview of CMA Platforms and Interpretation of Prenatal Examinations
4.2. Detection of Uniparental Disomy by CGH and SNP Platforms
4.3. Does CMA Provide a Higher Detection YIELD Than Karyotyping?
4.4. Does CMA Have a Higher Detection Yield for Fetuses with Increased NT and cFTS?
4.5. Relationship between CMA and Ultrasound Scan Abnormalities
4.6. CNVs and Advanced Maternal Age
4.7. Prenatal Clinical Counseling for Inherited CNVs or CNVs with Variable Penetrance and Expressivity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, B.; Wapner, R. Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 2018, 109, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, M.; Wang, H.; Guo, Y.; Chau, M.H.K.; Yan, H.; Cao, Y.; Kwok, Y.K.Y.; Chen, J.; Hui, A.S.Y.; et al. Clinical utility of expanded non-invasive prenatal screening and chromosomal microarray analysis in high-risk pregnancy. Ultrasound Obstet Gynecol. 2021, 57, 459–465. [Google Scholar] [CrossRef]
- South, S.T.; Lee, C.; Lamb, A.N.; Higgins, A.W.; Kearney, H.M. ACMG standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet. Med. 2013, 15, 901–909. [Google Scholar] [CrossRef]
- Vogel, I.; Petersen, O.B.; Christensen, R.; Hyett, J.; Lou, S.; Vestergaard, E.M. Chromosomal microarray as primary diagnostic genomic tool for pregnancies at increased risk within a population-based combined first-trimester screening program. Ultrasound Obstet. Gynecol. 2018, 51, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Hu, T.; Wang, J.M.; Li, Q.Q.; Wang, H.; Liu, S.L. Prenatal Diagnostic Value of Chromosomal Microarray in Fetuses with Nuchal Translucency Greater than 2.5 mm. BioMed Res. Int. 2019, 2019, 6504159. [Google Scholar] [CrossRef] [Green Version]
- Mademont-Soler, I.; Morales, C.; Soler, A.; Mart´ınez-Crespo, J.M.; Shen, Y.; Margarit, E.; Clusellas, N.; Obo’n, M.; Wu, B.L.; Sa´nchez, A. Prenatal diagnosis of chromosomal abnormalities in fetuses with abnormal cardiac ultrasound findings: Evaluation of chromosomal microarray-based analysis. Ultrasound Obstet. Gynecol. 2013, 41, 375–382. [Google Scholar] [CrossRef]
- Zhu, X.; Li, J.; Ru, T.; Wang, Y.; Xu, Y.; Yang, Y.; Wu, X.; Cram, D.S.; Hu, Y. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat. Diagn. 2016, 36, 321–327. [Google Scholar] [CrossRef]
- Charlotte, B.A.; Behar, D.M.; Herrero, S.G.; Rubio, C. Overcoming Challenges in Reproductive Health Applications by Deploying More Sensitive and Accurate Molecular Technologies. EMJ Repro. Health 2019, 5 (Suppl. S1), 2–12. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde MLyon, E.; Spector, E.; Voelkerding, K.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RooneyRiggs, E.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of 451 constitutional copy-number variants: A joint consensus recommendation of the American College of Medical 452 Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet. Med. 2013, 15, 478–481. [Google Scholar] [CrossRef]
- Hillman, S.; McMullan, D.J.; Maher, E.R.; Kilby, M.D. The use of chromosomal microarray in prenatal diagnosis. Obstet. Gynaecol. 2013, 15, 80–84. [Google Scholar] [CrossRef]
- De Wit, M.C.; Srebniak, M.I.; Govaerts, L.C.P.; Van Opstal, D.; Galjaard, R.J.H.; Go, A.T.J.I. Additional value of prenatal genomic array testing in fetuses with isolated structural ultrasound abnormalities and a normal karyotype: A systematic review of the literature. Ultrasound Obstet. Gynecol. 2014, 43, 139–146. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Rosenfeld, J.A.; Dabell, M.P.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Fisher, A.J. Detection rates of clinically significant genomic alterations by microarray analysis for specific anomalies detected by ultrasound. Prenat. Diagn. 2012, 32, 986–995. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, J.C.; Platt, L.P.; Rebarber, A.; Zachary, J.; Grobman, W.A.; Wapner, R.J. Association of Copy Number Variants with Specific Ultrasonographically Detected Fetal Anomalies. Obstet. Gynecol. 2014, 124, 83–90. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists Committee on Genetics. Committee opinion no. 682: Microarray and Next-Generation Sequencing Technology: The use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology. Obstet. Gynecol. 2016, 128, e262–e268. [Google Scholar] [CrossRef] [PubMed]
- Armour, C.M.; Dougan, S.D.; Brock, J.A.; Chari, R.; Chodirker, B.N.; DeBie, I.; Evans, J.A.; Gibson, W.T.; Kolomietz, E.; Nelson, T.N.; et al. Practice guideline: Joint CCMG-SOGC recommendations for the use of chromosomal microarray analysis for prenatal diagnosis and assessment of fetal loss in Canada. J. Med. Genet. 2018, 55, 215–221. [Google Scholar] [CrossRef]
- Gardiner, C.; Wellesley, D.; Kilby, M.D.; Kerr, B. Recommendations for the Use of Chromosome Microarray in Pregnancy. 2015, Volume 2015, pp. 1–17. Available online: https://www.rcpath.org/resourceLibrary/recommendations-for-the-use-of-chromosome-microarray-inpregnancy.html (accessed on 1 March 2022).
- Coe, B.P.; Witherspoon, K.; A Rosenfeld, J.; Van Bon, B.W.M.; Silfhout, A.T.V.-V.; Bosco, P.; Friend, K.L.; Baker, C.; Buono, S.; Vissers, L.; et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 2014, 46, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Oneda, B.; Baldinger, R.; Reissmann, R.; Reshetnikova, I.; Krejci, P.; Masood, R.; Ochsenbein-Kölble, N.; Bartholdi, D.; Steindl, K.; Morotti, D. High-resolution chromosomal microarrays in prenatal diagnosis significantly increase diagnostic power. Prenat. Diagn. 2014, 34, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Egloff, M.; Hervé, B.; Quibel, T.; Jaillard, S.; Le Bouar, G.; Uguen, K.; Saliou, A.-H.; Valduga, M.; Perdriolle, E.; Coutton, C. Diagnostic yield of chromosomal microarray analysis in fetuses with isolated increased nuchal translucency: A French multicenter study. Ultrasound Obstet. Gynecol. 2018, 52, 715–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagi-Dain, L.; Maya, I.; Reches, A.; Frumkin, A.; Grinshpun-Cohen, J.; Segel, R.; Manor, E.; Khayat, M.; Tenne, T.; Banne, E.; et al. Chromosomal Microarray Analysis Results from Pregnancies with Various Ultrasonographic Anomalies. Obstet. Gynecol. 2018, 132, 1368–1375. [Google Scholar] [CrossRef]
- Shi, Y.; Ma, J.; Xue, Y.; Wang, J.; Yu, B.; Wang, T. The assessment of combined karyotype analysis and chromosomal microarray in pregnant women of advanced maternal age: A multicenter study. Ann. Transl. Med. 2019, 7, 318. [Google Scholar] [CrossRef]
- Wang, J.C.; Radcliff, J.; Coe, S.J.; Mahon, L.W. Effects of platforms, size filter cutoffs, and targeted regions of cytogenomic microarray on detection of copy number variants and uniparental disomy in prenatal diagnosis: Results from 5026 pregnancies. Prenat. Diagn. 2019, 39, 137–156. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.U.; Jong, Y.J.; Huang, P.C.; Tsai, C. Detection of copy number variants with chromosomal microarray in 10 377 pregnancies at a single laboratory. Acta Obstet. Gynecol. Scand. 2020, 99, 775–782. [Google Scholar] [CrossRef]
- Xia, M.J.; Yang, X.; Fu, J.; Teng, Z.; Lv, Y.; Yu, L. Application of chromosome microarray analysis in prenatal diagnosis. BMC Pregnancy Childbirth 2020, 20, 696. [Google Scholar] [CrossRef]
- Hu, T.; Tian, T.; Zhang, Z.; Wang, J.M.; Hu, R.; Xiao, L.; Zhu, H.M.; Lai, Y.; Wang, H.; Liu, S.L. Prenatal chromosomal microarray analysis in 2466 fetuses with ultrasonographic soft markers: A prospective cohort study. Am. J. Obstet. Gynecol. 2021, 224, e1–e516. [Google Scholar] [CrossRef]
- Hu, Z.M.; Li, L.L.; Zhang, H.; Zhang, H.G.; Liu, R.Z.; Yu, Y. Clinical Application of Chromosomal Microarray Analysis in Pregnant Women with Advanced Maternal Age and Fetuses with Ultrasonographic Soft Markers. Med. Sci. Monit. 2021, 27, e929074-e1. [Google Scholar] [CrossRef]
- Stern, S.; Hacohen, N.; Meiner, V.; Yagel, S.; Zenvirt, S.; Shkedi-Rafid, S.; Macarov, M.; Valsky, D.V.; Porat, S.; Yanai, N.; et al. Universal chromosomal microarray analysis reveals high proportion of copy number variants in low risk pregnancies. Ultrasound Obstet. Gynecol. 2021, 57, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Q.; Li, Y.; Lin, N.; Xie, X.R.; Su, L.; Cai, M.Y.; Lin, Y.; Wang, L.S.; Wang, M.Y.; Xu, L.P.; et al. Chromosomal microarray analysis for pregnancies with abnormal maternal serum screening who undergo invasive prenatal testing. J. Cell Mol. Med. 2021, 25, 6271–6279. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, L.G.; Agan, N.; Goldberg, J.D.; Ledbetter, D.H.; Longshore, J.W.; Cassidy, S.B. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet. Med. 2001, 3, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudio, D.D.; Shinawi, M.; Astbury, C.; Tayeh, M.K.; Deak, K.L.; Raca, G.; ACMG Laboratory Quality Assurance Committee. Diagnostic testing for uniparental disomy: A points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1133–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grande, M.; Jansen, F.A.R.; Blumenfeld, Y.J.; Fisher, A.; Odibo, A.O.; Haak, M.C.; Borrell, A. Genomic microarray in fetuses with increased nuchal translucency and normal karyotype: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2015, 46, 650–658. [Google Scholar] [CrossRef]
- Lund, I.C.B.; Christensen, R.; Petersen, O.B.; Vogel, I.; Vestergaard, E.M. Chromosomal microarray in fetuses with increased nuchal translucency. Ultrasound Obstet. Gynecol. 2015, 45, 95–100. [Google Scholar] [CrossRef]
- Jansen, F.A.R.; Blumenffld, Y.J.; Fisher, A.; Cobben, J.M.; Odibo, A.O.; Borrell, A.; Haak, M.C. Array comparative genomic hybridization and fetal congenital heart defects: A systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2015, 45, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Landstrom, A.P.; Kim, J.J.; Gelb, B.D.; Helm, B.M.; Kannankeril, P.J.; Semsarian, C.; Sturm, A.C.; Tristani-Firouzi, M.; Ware, S.M. Genetic Testing for Heritable Cardiovascular Diseases in Pediatric Patients. Circ. Genom. Precis. Med. 2021, 14, e000086. [Google Scholar] [CrossRef]
- Zaidi, S.; Brueckner, M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017, 120, 923–940. [Google Scholar] [CrossRef]
- Maya, I.; Sharony, R.; Yacobson, R.; Kahana, S.; Yeshaya, J.; Tenne, T.; Agmon-Fishman, T.; Cohen-Vig, L.; Goldberg, Y.; Berger, R.; et al. When genotype is not predictive of phenotype: Implications for genetic counseling based on 21,594 chromosomal microarray analysis examinations. Genet. Med. 2018, 20, 128–131. [Google Scholar] [CrossRef] [Green Version]
- Rosias, P.R.; Sijstermans, J.M.; Theunissen, P.M.; Pulles-Heintzberger, C.F.; De Die-Smulders, C.E.; Engelen, J.J.; Van Der Meer, S.B. Phenotypic variability of the cat eye syndrome. Case report and review of the literature. Genet. Couns. 2001, 12, 273–282. [Google Scholar] [PubMed]
- Meins, M.; Burfeind, P.; Motsch, S.; Trappe, R.; Burtmus, D.; Langer, S.; Speicher, M.R.; Mühlendyck, H.; Bartels, I.; Zoll, B. Partial trisomy of chromosome 22 resulting from an interstitial duplication of 22q11.2 in a child with typical cat eye syndrome. J. Med. Genet. 2003, 40, e62. Available online: http://www.jmedgenet.com/cgi/content/full/40/5/e62 (accessed on 1 March 2022). [CrossRef] [PubMed] [Green Version]
- Crawford, K.; Bracher-Smith, M.; Owen, D.; Kendall, K.M.; Rees, E.; Pardiñas Einon, M.; Escott-Price, V.; Walters, J.T.R.; O’Donovan, M.C.; Owen, M.J.; et al. Medical consequences of pathogenic CNVs in adults: Analysis of the UK Biobank. J. Med. Genet. 2019, 56, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Silber, S.J. Y Chromosome Infertility 1993–2022; University of Washington: Seattle, WA, USA, 2002; ISSN 2372-0697. [Google Scholar]
- Bernhardt, B.A.; Soucier, D.; Hanson, K.; Savage, M.S.; Jackson, L.; Wapner, R.J. Women’s experiences receiving abnormal prenatal chromosomal microarray testing results. Genet. Med. 2013, 15, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, L.; Akkari, Y.; Cooley, L.D.; Miller, D.T.; Seifert, B.A.; Wolff, D.J.; Mikhail, F.M.; ACMG Laboratory Quality Assurance Committee. Chromosomal microarray analysis, including constitutional and neoplastic disease applications, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1818–1829. [Google Scholar] [CrossRef]

{kind=link}
| Indication | No | T21 | T18 | T13 | Reported CNVs ≥ 400 kb | Nonreported CNVs ≥ 400 kb |
|---|---|---|---|---|---|---|
| Advanced maternal age only | 546 (52.7%) | 1 | 1 | 3 | 3 | 14 |
| Abnormal ultrasound finding | 197 (19.0%) | 2 | 4 | 1 | 11 | 2 |
| High risk on Down syndrome screening (≥1:270) | 189 (18.2%) | 13 | 10 | 3 | 13 | 9 |
| Increased NT (≥3.5 mm) only | 90 | 6 | 5 | 2 | 1 | 3 |
| FTS soft markers: DV(+), TR(+), NB(−) | 56 | 7 | 5 | 1 | 1 | 4 |
| Previous pregnancy, child, or familial risk | 105 (10.1%) | 1 | 5 |
| Case | Age | NT (mm) | Indication/Ultrasound Finding | CMA [hg19] Results | Size (Mb) | Inheritance | Candidate OMIM Genes | Disorders | Interpretation | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 34 | 2.5 | TR(+), High risk | 1q21.1q21.2 (146,627,038–147,384,032) × 1 | 0.757 | De novo | 612474 | 1q21.1 deletion syndrome | Pathogenic | TOP |
| 2 | 35 | 3.5 | NT(+), High risk | 2q37.2q37.3 (236,330,093–243,040,324) × 1 | 6.710 | De novo | 600430 | 2q37 deletion syndrome | Pathogenic | TOP |
| 3 | 36 | 3.2 | Omphalocele, High risk | 3p26.33p26.2 (2,146,782–3,771,742) × 3 | 1.625 | Paternal | 607280 | 3pter-p25 duplication | VOUS | TOP |
| 4 | 21 | 2.2 | CM(+), TR(+), NB(−),High risk | 3q22.1q25.32 (130,521,560–157,015,801) × 1 | 26.494 | De novo | 220200 | Syndromic intellectual disability Dandy Walker Syndrome | Pathogenic | TOP |
| 5 | 38 | 5.1 | NT(+), DV(+), High risk, AMA | 3q27.2q29 (184,799,629–197,803,820) × 3 9p24.3p22.3 (271,257–14,680,180) × 1 9p22.3p13.1 (14,844,795–38,663,271) × 3 | 13.004 14.409 23.818 | De novo | 611936/602424/604935/612900/158170/608980/156540/601673 | 3q29 duplication syndrome 9p24.3p22 deletion 9p22.3p13.1 duplication 9p duplication & deletion | LP Pathogenic | TOP |
| 6 | 27 | 2.8 | High risk | 4q34.3q35.1 (180,742,112–183,532,267) × 3 4q35.1q35.2 (183,532,267–190,957,460) × 1 | 2.790 7.425 | De novo | 610083/518900 | 4q34.3q35.1 duplication 4q35.1q35.2 deletion | LP | TOP |
| 7 | 25 | 4 | Micrognathia, Low set ear, NT(+) | 5q32 (145,755,389–150,297,954) × 1 | 2.575 | De novo | N/A | Treacher Collins syndrome | Pathogenic | TOP |
| 8 | 36 | 2.8 | TR(+), High Risk, AMA | 6q22.1q22.31 (115,853,923–119,245,348) × 3 | 3.391 | De novo | 605942/604714/612647/612659/610463/618865/172405/610098/120110 | 6q22.1q22.31 duplication | VOUS | LB (MR) |
| 9 | 37 | 2 | AMA | 7q21.11q21.3 (84,600,949–96,051,291) × 1 | 11.45 | De novo | 600028/604149 | Split-hand/foot malformation 1 Silver–Russell syndrome Myoclonus-Dystonia | Pathogenic | TOP |
| 10 | 36 | 2.6 | High risk, AMA | 8q23.1 (106,336,068–106,715,982) × 1 | 0.380 | 603693 | congenital diaphragmatic hernia | LP | BH | |
| 11 | 35 | 5.1 | NT(+), AMA | 9p24.3p22.2 (204,193–16,626,507) × 1 | 16.422 | De novo | 158170 | 9p24.3p22.2 deletion syndrome | Pathogenic | TOP |
| 12 | 31 | 5.7 | Radial aplasia, NT(+), High risk | 9q21.2 (80,191,465–80,601,045) × 3 | 0.41 | De novo | 600998 | Radial aplasia | VOUS | TOP |
| 13 | 36 | 1.9 | High risk, AMA | 10q22.3 (79,617,635–81,707,527) × 3 | 2.090 | De novo | 602412/614258/607159/178642/178630/618639 | 10q22.3 duplication | VOUS | BH |
| 14 | 32 | 1.4 | Double outlet of right ventricle, TR(+) | 10q23.1 (86,767,729–86,984,308) × 1 | 0.217 | De novo | N/A | 10q23 deletion syndrome | VOUS | TOP |
| 15 | 35 | 4.5 | NT(+), High risk, AMA | 14q32.31q32.33 (101,758,166–106,852,173) × 1 | 5.094 | De novo | 614062/605799/614730 | 14q32.31q32.33 deletion syndrome | Pathogenic | TOP |
| 16 | 27 | 1.9 | TOF of Heart, NB(−), High risk | 16p11.2 (29,653,115–30,198,522) × 1 | 0.545 | De novo | 611913 | Proximal 16p11.2 deletion syndrome | Pathogenic | TOP |
| 17 | 31 | 4.3 | R/O: VACTERL, NT(+), High risk | 16p11.2 (29,653,115–30,198,581) × 1 | 0.545 | De novo | 611913 | Proximal 16p11.2 deletion syndrome | Pathogenic | TOP |
| 18 | 32 | 1.5 | Megacystis | 16p11.2 (29,698,283–30,198,582) × 3 | 0.500 | De novo | 614671 | Proximal 16p11.2 duplication syndrome | Pathogenic | BH |
| 19 | 37 | 2.3 | TR(+), NB(−), High risk | 16p13.11 (15,131,575–16,288,874) × 3 | 1.157 | Maternal | 609449/160745/603234 | 16p13.1 duplication | LP | IUFD |
| 20 | 40 | 1.7 | Pyelectasis | 22q11.1q11.21 (17,444,646–18,106,018) × 3 | 0.661 | De novo | 115470 | Cat eye syndrome (47,XY+ mar de novo [42]/46,XY [29]) | Pathogenic | TOP |
| 21 | 37 | 2 | AMA | 22q11.1q11.21 (17,444,646–17,993,089) × 3 | 0.548 | Maternal | 115470 | 22q11.1q11.21 duplication (46,XY) | LP | BH |
| 22 | 31 | 9.4 | NT(+), NB(−), TR(+), High risk | 22q11.21 (19,035,231–21,449,413) × 1 | 2.414 | De novo | 188400 | DiGeorge Syndrome | Pathogenic | TOF |
| 23 | 42 | 4.3 | NT(+), TR(+), High risk, AMA | 22q11.21 (19,006,943–21,461,068) × 1 | 2.454 | De novo | 188400 | DiGeorge syndrome | Pathogenic | TOP |
| 24 | 42 | 2.5 | TR(+), High risk, AMA | 22q11.21 (19,006,943–21,461,005) × 1 17q12 (34,823,708–36,247,940) × 3 | 2.454 1.426 | De novo | 614526 188400 | DiGeorge syndrome 17q12 duplication syndrome | Pathogenic | TOP |
| 25 | 33 | 4.4 | NT(+) | 22q11.21 (18,104,691–21,461,005) × 3 | 3.356 | De novo | 608363 | 22q11.2 duplication syndrome | Pathogenic | TOP |
| 26 | 35 | 2.1 | PHx; Genetic Hx | 22q11.21 (19,006,943–21,461,005) × 3 | 2.454 | Maternal | 608363 | 22q11.2 duplication syndrome | Pathogenic | BH (Health) |
| 27 | 40 | 2.3 | AMA | Xp22.31 (6,460,120–8,101,239) × 1 | 1.641 | De novo | 308100 | X-linked mental retardation Ichthyosis, X-linked (XLI) | Pathogenic | TOP |
| 28 | 31 | 1.4 | NB(−), Mental retardation | Xq22.1q22.2 (100,907,854–102,659,284) × 1 | 1.751 | De novo | 300319/300969 | Xq22.1q22.2 deletion X-linked mental retardation | LP | LB (MR) |
| 29 | 29 | 2.1 | Ambigious genital | Xq28 (154,130,347–154,527,746) × 3 | 0.397 | De novo | 300815 | Xq28 duplication syndrome | Pathogenic | TOP |
| 30 | 31 | 2.5 | Echogenic bowel | Xq28 (154,161,678–154,650,677) × 3 7q36.2 (153,923,581–154,024,097) × 1 | 0.351 0.087 | Paternal Maternal | 300815 612956 | Xq28 duplication syndrome 7q36.2 deletion syndrome | Pathogenic VOUS | BH |
| Author | Patient Population | Cases No | CMA Platform | Chip Design | CMA Resolution | Interpretation Cut Off | Detection Rate | P/LP CNVs |
|---|---|---|---|---|---|---|---|---|
| Oneda et al. (2014) [23] | High risk # | 464 | Affymetrix cytogenetics Whole Genome 2.7 M array/Cytoscan HD Array | CGH + SNP | 20–100 Kb | 20–100 Kb | 17/464 (3.70%) | 15/464 (3.23%) |
| Zhu et al. (2016) [5] | Heart anomaly | 115 | Affymetrix CytoScan 750 K | CGH + SNP | 100 Kb | N/A | 21/115 (18.3%) | 13/115 (11.3%) |
| Egloff et al. (2018) [24] | High risk # | 599 | Agilent PreCytoNEM | CGH + SNP | 60 &180 Kb | N/A | 53/599 (8.85%) | 16/599 (2.67%) |
| Sagi-Dain et al. (2018) [25] | Ultrasound anomaly | 5750 | Affymetrix CytoScan 750 K array Infinium OmniExpress-24 v1.2 BeadChip BlueGnome Cytochip ISCA 8 × 60 K format Agilent CGH + SNP (4 × 180 K) | CGH + SNP SNP CGH CGH + SNP | 100 Kb | 1 M (loss)/2 M (gain) | 272/5750 (4.73%) | 157/5750 (2.73%) |
| Vogel et al. (2018) [7] | cFTS high risk | 575 | Agilent CytoGenomics | CGH + SNP | 180 K | N/A | 51/575 (8.87%) | 15/575 (2.61%) |
| Shi et al. (2019) [26] | High risk, AMA | 703 | Affymetrix CytoScan 750 K | CGH + SNP | 100 Kb | N/A | 48/703 (6.83%) | 10/703 (1.42%) |
| Wang et al. (2019) [27] | High risk # | 5026 | Affymetrix Human SNP Array 6.0 Affymetrix CytoScan HD | SNP CGH + SNP | Target: 20 kb (loss)/100 kb (gain) Nontarget: 50 kb (loss)/200 kb (gain) | 400 K | 562/5026 (11.2%) | 19/5026 (0.38%) |
| Lin et al. (2020) [28] | General population | 10,377 | Thermo-Fisher CytoScan750 K | CGH + SNP | 100 Kb | 200 K | 223/10,377 (2.15%) | 126/10,377 (1.21%) |
| Xia et al. (2020) [29] | Ultrasound anomaly | 477 | Affymetrix CytoScan 750 K | CGH + SNP | 50 Kb (loss)/100 Kb (gain) | 100 K (loss)/200 K (gain) | 71/447 (15.88%) | 17/447 (3.80%) |
| Hu et al. (2021) [30] | Ultrasound anomaly | 2466 | Thermo-Fisher CytoScan750 K | CGH + SNP | 100 Kb | 400 K | 107/2466 (4.34%) | 64/2466 (2.59%) |
| Hu et al. (2021) [31] | AMA, soft marker | 1521 | Affymetrix CytoScan 750 K | CGH + SNP | 100 Kb | 400 K | 330/1527 (21.61%) | 37/1520 (2.42%) |
| Stern et al. (2021) [32] | Ultrasound low risk | 6431 | Affymetrix CytoScan 750 K | CGH + SNP | 100 Kb | N/A | 319/6431 (4.96%) | 27/6431 (0.42%) |
| Wu et al. (2021) [33] | Serum screening high risk | 713 | Affymetrix CytoScan 750 K | CGH + SNP | 100 Kb | 400 K | 82/713 (11.5%) | 59/713 (8.27%) |
| Zhu et al. (2021) [10] | High risk # | 774 | Affymetrix CytoScan 750 K Iliumina HumanCytoSNP-12 Agilent CGH 8 × 60 K (customized) | CGH + SNP SNP CGH + SNP | 100 Kb | 400 K | 308/774 (39.79%) | 17/774 (2.20%) |
| Present study | High risk # | 1037 | Phalanx CytoOne | CGH | Target: 50–100 K; Non-target: 1 Mb | 400 K | 153/1037 (14.75%) | 30/1037 (2.89%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsiao, C.-H.; Chen, J.-S.; Shiao, Y.-M.; Chen, Y.-J.; Chen, C.-H.; Chu, W.-C.; Wu, Y.-C. Prenatal Diagnosis Using Chromosomal Microarray Analysis in High-Risk Pregnancies. J. Clin. Med. 2022, 11, 3624. https://doi.org/10.3390/jcm11133624
Hsiao C-H, Chen J-S, Shiao Y-M, Chen Y-J, Chen C-H, Chu W-C, Wu Y-C. Prenatal Diagnosis Using Chromosomal Microarray Analysis in High-Risk Pregnancies. Journal of Clinical Medicine. 2022; 11(13):3624. https://doi.org/10.3390/jcm11133624
Chicago/Turabian StyleHsiao, Ching-Hua, Jia-Shing Chen, Yu-Ming Shiao, Yann-Jang Chen, Ching-Hsuan Chen, Woei-Chyn Chu, and Yi-Cheng Wu. 2022. "Prenatal Diagnosis Using Chromosomal Microarray Analysis in High-Risk Pregnancies" Journal of Clinical Medicine 11, no. 13: 3624. https://doi.org/10.3390/jcm11133624
APA StyleHsiao, C.-H., Chen, J.-S., Shiao, Y.-M., Chen, Y.-J., Chen, C.-H., Chu, W.-C., & Wu, Y.-C. (2022). Prenatal Diagnosis Using Chromosomal Microarray Analysis in High-Risk Pregnancies. Journal of Clinical Medicine, 11(13), 3624. https://doi.org/10.3390/jcm11133624