
A Genotype-Phenotype Study of Multiple Hereditary Exostoses in Forty-Three Patients


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
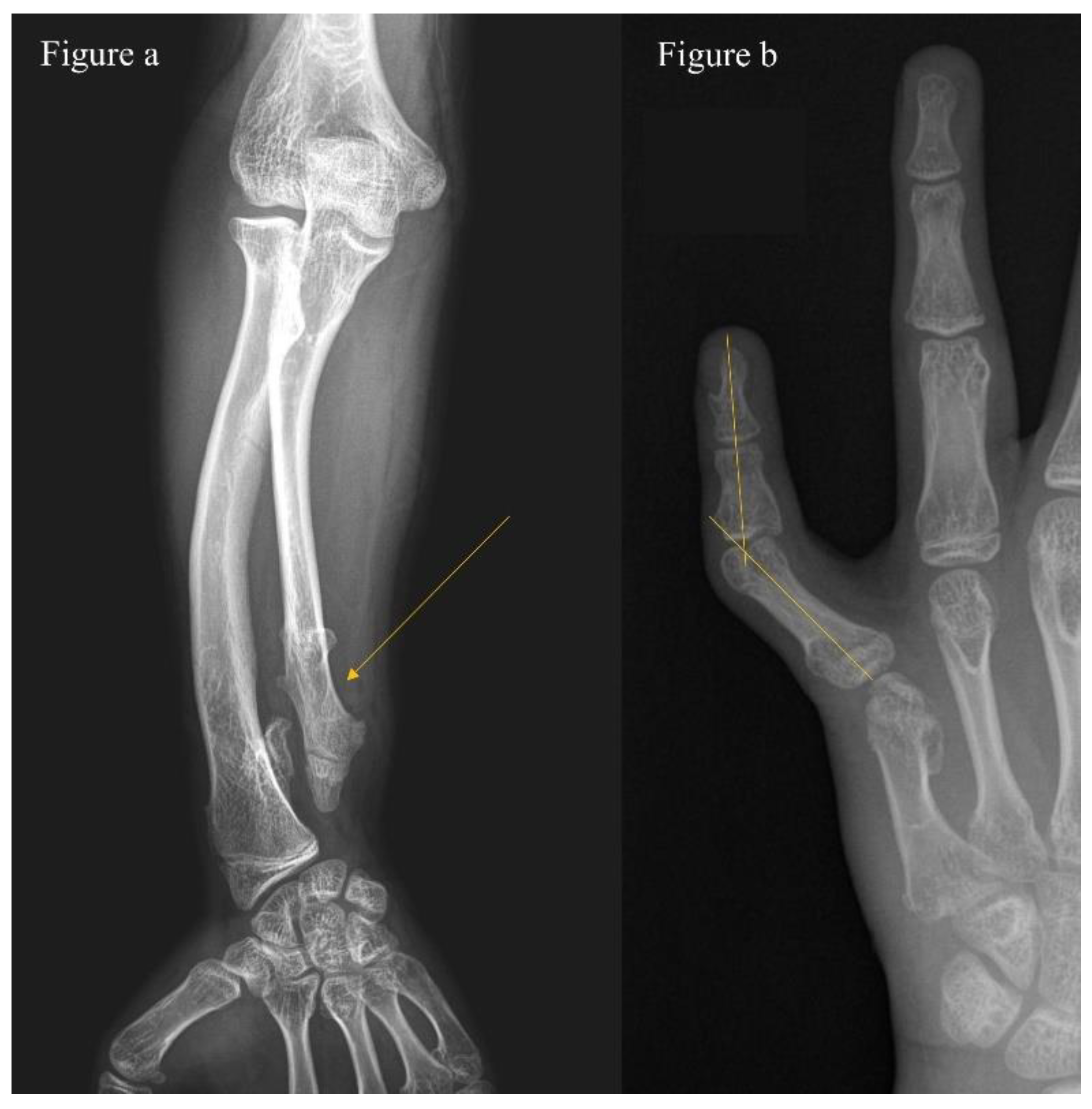
2.2. Clinical Phenotype Study and Classification System
2.3. Molecular Screening
2.4. Statistics
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jamsheer, A.; Socha, M.; Sowińska-Seidler, A.; Telega, K.; Trzeciak, T.; Latos-Bieleńska, A. Mutational screening of EXT1 and EXT2 genes in Polish patients with hereditary multiple exostoses. J. Appl. Genet. 2014, 55, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Stieber, J.R.; Dormans, J.P. Manifestations of Hereditary Multiple Exostoses. J. Am. Acad. Orthop. Surg. 2005, 13, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Hennekam, R.C. Hereditary multiple exostoses. J. Med. Genet. 1991, 28, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicklund, C.L.; Pauli, R.M.; Johnston, D.; Hecht, J.T. Natural history study of hereditary multiple exostoses. Am. J. Med Genet. 1995, 55, 43–46. [Google Scholar] [CrossRef]
- McCormick, C.; Duncan, G.; Goutsos, K.T.; Tufaro, F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc. Natl. Acad. Sci. USA 2000, 97, 668–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busse, M.; Feta, A.; Presto, J.; Wilén, M.; Grønning, M.; Kjellén, L.; Kusche-Gullberg, M. Contribution of EXT1, EXT2, and EXTL3 to Heparan Sulfate Chain Elongation. J. Biol. Chem. 2007, 282, 32802–32810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Lüdecke, H.-J.; Lindow, S.; Horton, W.A.; Lee, B.; Wagner, M.J.; Horsthemke, B.; Wells, D.E. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nat. Genet. 1995, 11, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.; Van Hul, W.; Wauters, J.; Nemtsova, M.; Reyniers, E.; Van Hul, E.; De Boulle, K.; De Vries, B.B.A.; Hendrickx, J.; Herrygers, I.; et al. Positional cloning of a gene involved in hereditary multiple exostoses. Hum. Mol. Genet. 1996, 5, 1547–1557. [Google Scholar] [CrossRef] [Green Version]
- Stickens, D.; Clines, G.; Burbee, D.; Ramos, P.; Thomas, S.; Hogue, D.; Hecht, J.T.; Lovett, M.; Evans, G.A. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nat. Genet. 1996, 14, 25–32. [Google Scholar] [CrossRef]
- Lind, T.; Tufaro, F.; McCormick, C.; Lindahl, U.; Lidholt, K. The Putative Tumor Suppressors EXT1 and EXT2 Are Glycosyltransferases Required for the Biosynthesis of Heparan Sulfate. J. Biol. Chem. 1998, 273, 26265–26268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, C.; Duncan, G.; Tufaro, F. New perspectives on the molecular basis of hereditary bone tumours. Mol. Med. Today 1999, 5, 481–486. [Google Scholar] [CrossRef]
- Bellaiche, Y.; The, I.; Perrimon, N. Tout-velu is a Drosophila homologue of the putative tumour suppressor EXT-1 and is needed for Hh diffusion. Nature 1998, 394, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.P.; Mitchell, J. Indian hedgehog: Its roles and regulation in endochondral bone development. J. Cell. Biochem. 2005, 96, 1163–1173. [Google Scholar] [CrossRef]
- Jennes, I.; Pedrini, E.; Zuntini, M.; Mordenti, M.; Balkassmi, S.; Asteggiano, C.G.; Casey, B.; Bakker, B.; Sangiorgi, L.; Wuyts, W. Multiple osteochondromas: Mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum. Mutat. 2009, 30, 1620–1627. [Google Scholar] [CrossRef]
- Francannet, C.; Cohen-Tanugi, A.; Le Merrer, M.; Munnich, A.; Bonaventure, J.; Legeai-Mallet, L. Genotype-phenotype correlation in hereditary multiple exostoses. J. Med Genet. 2001, 38, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Jäger, M.; Westhoff, B.; Portier, S.; Leube, B.; Hardt, K.; Royer-Pokora, B.; Goßheger, G.; Krauspe, R. Clinical outcome and genotype in patients with hereditary multiple exostoses. J. Orthop. Res. 2007, 25, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, J.; Wang, Z.; Tang, J.; Yu, T. A genotype-phenotype study of hereditary multiple exostoses in forty-six Chinese patients. BMC Med Genet. 2017, 18, 126. [Google Scholar] [CrossRef] [Green Version]
- Pedrini, E.; Jennes, I.; Tremosini, M.; Milanesi, A.; Mordenti, M.; Parra, A.; Sgariglia, F.; Zuntini, M.; Campanacci, L.; Fabbri, N.; et al. Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: Identification of “protective” and “risk” factors. J. Bone Jt. Surg. Am. 2011, 93, 2294–2302. [Google Scholar] [CrossRef]
- Alvarez, C.; Tredwell, S.; De Vera, M.; Hayden, M. The genotype-phenotype correlation of hereditary multiple exostoses. Clin. Genet. 2006, 70, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, W.; Van Hul, W. Molecular basis of multiple exostoses: Mutations in the EXT1 and EXT2 genes. Hum. Mutat. 2000, 15, 220–227. [Google Scholar] [CrossRef]
- Malini, K.; Gudi, N.S.; Kutty, A.V.M.; Balakrishna, S. Mutational Analysis of Exostosin 1 and 2 Genes in Multiple Osteochondroma. Indian J. Pediatr. 2015, 82, 649–650. [Google Scholar] [CrossRef]
- Hecht, J.T.; Hogue, D.; Wang, Y.; Blanton, S.H.; Wagner, M.; Strong, L.C.; Raskind, W.; Hansen, M.F.; Wells, D. Hereditary multiple exostoses (EXT): Mutational studies of familial EXT1 cases and EXT-associated malignancies. Am J Hum Genet 1997, 60, 80–86. [Google Scholar] [PubMed]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Schmale, G.A.; Conrad, E.U., 3rd; Raskind, W.H. The natural history of hereditary multiple exostoses. J. Bone Jt. Surg. Am. 1994, 76, 986–992. [Google Scholar] [CrossRef]
- Sarrión, P.; Sangorrin, A.; Urreizti, R.; Delgado, M.A.; Artuch, R.; Martorell, L.; Armstrong, J.; Anton, J.; Torner, F.; Vilaseca, M.A.; et al. Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Sci. Rep. 2013, 3, 1346. [Google Scholar] [CrossRef] [Green Version]
- Signori, E.; Massi, E.; Matera, M.G.; Poscente, M.; Gravina, C.; Falcone, G.; Rosa, M.A.; Rinaldi, M.; Wuyts, W.; Seripa, D.; et al. A combined analytical approach reveals novel ext1/2 gene mutations in a large cohort of italian multiple osteochondromas patients. Genes Chromosomes Cancer 2007, 46, 470–477. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Score |
|---|---|
| Deformity | Maximum of 4 |
| Upper (except hand/ elbow dislocation = 1) | 0.5 (1) |
| Lower (except foot) | 0.5 |
| Hand | 0.5 |
| Foot | 0.5 |
| Additional | |
| Both | 0.5 |
| Hip and knee and ankle | 0.5 |
| Number of masses | Maximum of 2 |
| ≥20 | |
| 10≤ <20 | 1 |
| <10 | |
| LLD | Maximum of 2 |
| ≥20 | 2 |
| 10≤ <20 | 1 |
| Function | Maximum of 2 |
| Pain | |
| Limited ROM |
| EXT1 n = 21 | EXT2 n = 18 | No Mutation n = 4 | p-Value | EXT1 vs. EXT2 | EXT 1 vs. No Mutation | EXT2 vs. No Mutation | |
|---|---|---|---|---|---|---|---|
| Total clinical score Mean (±SD) | 5.76 (±1.60) | 4.06 (±1.47) | 4.63 (±1.44) | 0.005 1 | 0.004 | 0.544 | >1.0 |
| Deformity score Mean (±SD) | 1.86 (±0.88) | 0.94 (±0.84) | 1.13 (±0.48) | 0.018 1 | 0.004 | 0.367 | >1.0 |
| Number of exostoses Mean (±SD), range | 29.19 (±8.18), 12–52 | 20.11 (±8.18), 7–39 | 21.75 (±9.91), 7–28 | 0.018 1 | 0.017 | 0.499 | >1.0 |
| Severity | 0.011 2 | ||||||
| Mild | 2 (9.5%) | 8 (44.4%) | 1 (25.0%) | – | – | – | |
| Moderate | 7 (33.3%) | 7 (38.9%) | 3 (75.0%) | – | – | – | |
| Severe | 12 (57.1%) | 3 (16.7%) | 0 (0.0%) | – | – | – | |
| Gender | 0.814 2 | ||||||
| Male | 9 (42.9%) | 10 (55.6%) | 2 (50.0%) | – | – | – | |
| Female | 12 (57.1%) | 8 (44.4%) | 2 (50.0%) | – | – | – | |
| Familial or sporadic | 0.066 2 | ||||||
| Sporadic | 2 (9.5%) | 0 (0.0%) | 2 (28.6%) | – | – | – | |
| Familial | 18 (85.7%) | 17 (94.4%) | 2 (71.4%) | – | – | – | |
| Unknown | 1 (4.8%) | 1 (5.6%) | 0 (0.0%) | – | – | – | |
| Mutation variant | 0.076 2 | ||||||
| Nonsense | 1 (4.8%) | 4 (22.2%) | – | – | – | – | |
| Missense | 4 (19.0%) | 2 (11.1%) | – | – | – | – | |
| Frameshift | 8 (38.1%) | 6 (33.3%) | – | – | – | – | |
| Splice-site | 0 (0.0%) | 1 (5.6%) | – | – | – | – | |
| Big deletion or duplication |
| Patients | Family | Familial History | Sex | Clinical Class (Score) | EXT1 | EXT2 | Protein Change | Reference | Variant Type |
|---|---|---|---|---|---|---|---|---|---|
| 1 | A | f | M | Moderate (5.5) | c.1103delA | p.Glu368fs | Present study | Small deletion (Frameshift) | |
| 2 | B | f | M | Moderate (5.5) | c.1037 G>T | p.Arg346Ile | Jennes et al. [14] | Missense (likely pathogenic) | |
| 3 | B | f | F | Moderate (5.0) | c.1037 G>T | p.Arg346Ile | Jennes et al. [14] | Missense (likely pathogenic) | |
| 4 | B | f | M | Severe (7.0) | c.1037 G>T | p.Arg346Ile | Jennes et al. [14] | Missense (likely pathogenic) | |
| 5 | C | f | F | Severe (6.5) | E6, hetero deletion | Present study | Single exon deletion | ||
| 6 | D | f | F | Moderate (4.0) | E6-E8, hetero deletion | Present study | Multiple exon deletion | ||
| 7 | D | f | M | Mild (2.0) | E6-E8, hetero deletion | Present study | Multiple exon deletion | ||
| 8 | E | f | M | Mild (3.5) | c.610delG | p.Asp204fs | Present study | Small deletion (Frameshift) | |
| 9 | E | f | M | Moderate (4.0) | c.610delG | p.Asp204fs | Present study | Small deletion (Frameshift) | |
| 10 | F | f | F | Mild (3.0) | Whole gene, hetero deletion | Present study | EXT1 All Exon deletion | ||
| 11 | F | f | F | Severe (6.5) | Whole gene, hetero deletion | Present study | EXT1 All Exon deletion | ||
| 12 | F | f | F | Severe (8.0) | Whole gene, hetero deletion | Present study | EXT1 All Exon deletion | ||
| 13 | G | f | M | Severe (6.5) | c.112G>T | p.Glu38Ter | Present study | Nonsense | |
| 14 | H | f | F | Mild (3.0) | c.699T>G | p.Tyr233Ter | Wuyts et al. [20] | Nonsense | |
| 15 | H | f | M | Moderate (5.5) | c.699T>G | p.Tyr233Ter | Wuyts et al. [20] | Nonsense | |
| 16 | I | f | M | Mild (3.0) | c.67C>T, | p.Arg23Ter | Malini et al. [21] | Nonsense | |
| 17 | I | f | F | Moderate (5.0) | c.67C>T | p.Arg23Ter | Malini et al. [21] | Nonsense | |
| 18 | J | f | F | Mild (2.5) | No mutation | No mutation | – | – | |
| 19 | K | f | M | Moderate (5.0) | No mutation | No mutation | – | – | |
| 20 | L | s | F | Severe (6.5) | c.453delG | p.Ala151fs | Present study | Small deletion (Frameshift) | |
| 21 | M | f | M | Severe (6.0) | c.779dup | p.Gly259fs | Present study | Small duplication (Frameshift) | |
| 22 | N | f | M | Moderate (4.5) | E1-E2, triplicated | Present study | Exon Triplication | ||
| 23 | O | f | M | Severe (6.0) | c.1019G>T | p.Arg340Leu | Hecht et al. [22] | Missense (pathogenic) | |
| 24 | P | f | F | Moderate (4.0) | E1 heterodeletion | Present study | E1 heterodeletion | ||
| 25 | P | f | F | Severe (7.0) | E1 heterodeletion | Present study | E1 heterodeletion | ||
| 26 | P | f | F | Mild (2.0) | E1 heterodeletion | Present study | E1 heterodeletion | ||
| 27 | P | f | F | Severe (7.0) | E1 heterodeletion | Present study | E1 heterodeletion | ||
| 28 | R | f | F | Moderate (4.5) | E6-E8, heterodeletion | Present study | Multiple exon deletion | ||
| 29 | S | f | F | Moderate (4.5) | c.1103delA | p.Glu368fs | Present study | Small deletion (Frameshift) | |
| 30 | T | u | F | Moderate (4.0) | c.1469delT | p.Leu490fs | Ahn et al. [7] | Small deletion (Frameshift)9 | |
| 31 | U | s | M | Moderate (5.5) | No mutation | No mutation | – | – | |
| 32 | V | s | F | Moderate (5.5) | No mutation | No mutation | – | – | |
| 33 | W | f | F | Mild (2.0) | c.1345-1366del | c.484C>T | p.Gln162Ter | Nykamp et al. [23] | Nonsense |
| 34 | X | f | M | Moderate (5.0) | p.Pro449fs | Present study | Small deletion (frameshift) | ||
| 35 | Y | f | M | Severe (6.0) | c.536dupA | p.Gln179fs | Present study | Small duplication (Frameshift) | |
| 36 | Y | f | F | Severe (6.5) | c.536dupA | p.Gln179fs | Present study | Small duplication (Frameshift) | |
| 37 | Y | f | M | Severe (7.5) | c.536dupA | p.Gln179fs | Present study | Small duplication (Frameshift) | |
| 38 | Y | u | M | Severe (6.0) | c.1080-2del | Splice site | |||
| 39 | Z | s | M | Severe (8.0) | c.247dupC | p.Arg83fs | Small duplication (Frameshift) | ||
| 40 | A2 | f | M | Mild (2.5) | c.1103delA | pGlu368fs | Present study | Small deletion (Frameshift) | |
| 41 | B2 | f | F | Moderate (5.5) | c.89_95delCATCGAG | p.Ala30fs | Present study | Small deletion (Frameshift) | |
| 42 | C2 | f | F | Mild (2.0) | c.514C>T | p.Gln172Ter | Richards et al. [24] | Nonsense | |
| 43 | D2 | f | M | Mild (3.5) | E6-E8, heterodeletion | – | Present study | E2 heterodeletion |
| Sites | n (%) |
|---|---|
| Proximal humerus | 6 (15.8%) |
| Shaft of humerus | 26 (68.4%) |
| Distal humerus | 4 (10.5%) |
| Proximal Ulna | 9 (23.7%) |
| Distal Ulna | 30 (78.9%) |
| Proximal Radius | 11 (28.9%) |
| Distal Radius | 26 (68.4%) |
| Hand | 11 (28.9%) |
| Ilium | 17 (44.7%) |
| Ischium | 12 (31.6%) |
| Proximal Femur | 35 (92.1%) |
| Distal Femur | 38 (100%) |
| Proximal Tibia | 36 (94.7%) |
| Distal Tibia | 35 (92.1%) |
| Proximal Fibula | 34 (89.5%) |
| Distal Fibula | 16 (42.1%) |
| Foot | 8 (21.1%) |
| Upper Extremity | Lower Extremity | Short Bones | ||
|---|---|---|---|---|
| Hand | Foot | |||
| Involvement (a) | 40 | 43 | 14 | 9 |
| Bilateral involvement (b) | 11 | 24 | - | - |
| Proportion of both side involvement (b/a × 100) | 27.5% | 55.8% | - | - |
| Deformity (c) | 24 | 33 | 7 | 2 |
| Proportion of deformity (c/a × 100) | 60.0% | 76.74% | 50.0% | 22.22% |
| Factors | Univariate | Multivariate | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Odds Ratio | 95% CI | p-Value | Odds Ratio | 95% CI | p-Value | ||||
| Gene | EXT1 | Reference | - | - | - | ||||
| No | N/A | N/A | N/A | N/A | |||||
| EXT2 | 0.150 | 0.033 | 0.680 | 0.014 | 1.745 | 0.181 | 16.842 | 0.630 | |
| Sex | Male | Reference | - | - | - | ||||
| Female | 0.758 | 0.216 | 2.666 | 0.666 | |||||
| Familial history | Familial | 0.480 | 0.028 | 8.348 | 0.614 | ||||
| Sporadic | 1.000 | 0.034 | 29.807 | 1.000 | |||||
| Unknown | Reference | - | - | - | |||||
| Total number of exostoses | 1.206 | 1.071 | 1.358 | 0.002 | 1.084 | 0.943 | 1.246 | 0.259 | |
| Deformity score | 23.984 | 3.388 | 169.784 | 0.001 | 9.864 | 1.230 | 79.132 | 0.031 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Lee, C.-H.; Choi, S.-Y.; Kim, M.-K.; Jung, S.T. A Genotype-Phenotype Study of Multiple Hereditary Exostoses in Forty-Three Patients. J. Clin. Med. 2022, 11, 3703. https://doi.org/10.3390/jcm11133703
Kim S, Lee C-H, Choi S-Y, Kim M-K, Jung ST. A Genotype-Phenotype Study of Multiple Hereditary Exostoses in Forty-Three Patients. Journal of Clinical Medicine. 2022; 11(13):3703. https://doi.org/10.3390/jcm11133703
Chicago/Turabian StyleKim, Sungmin, Chang-Hyun Lee, Seok-Yong Choi, Myeong-Kyu Kim, and Sung Taek Jung. 2022. "A Genotype-Phenotype Study of Multiple Hereditary Exostoses in Forty-Three Patients" Journal of Clinical Medicine 11, no. 13: 3703. https://doi.org/10.3390/jcm11133703
APA StyleKim, S., Lee, C.-H., Choi, S.-Y., Kim, M.-K., & Jung, S. T. (2022). A Genotype-Phenotype Study of Multiple Hereditary Exostoses in Forty-Three Patients. Journal of Clinical Medicine, 11(13), 3703. https://doi.org/10.3390/jcm11133703